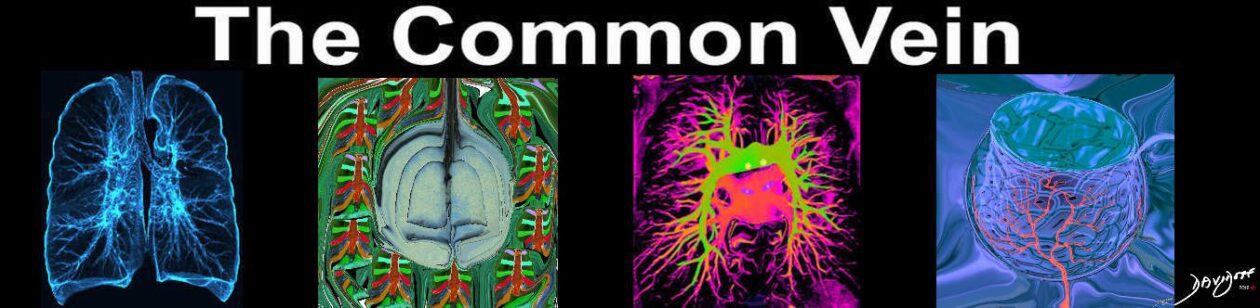
- Etymology:
The term “honeycomb lung” is derived from the characteristic appearance of clustered cystic spaces in the lungs resembling a honeycomb. - AKA:
Honeycombing, Honeycomb fibrosis (HCF). - What is it?
Honeycomb lung refers to the presence of clustered cystic airspaces in the subpleural regions of the lungs. These cysts are lined by bronchiolar epithelium, which is a hallmark of honeycombing at pathology, and represent end-stage pulmonary fibrosis. Despite being lined by bronchiolar epithelium, these cystic spaces are not connected to the bronchial tree due to surrounding fibrosis, which disrupts normal airway continuity. Honeycombing reflects irreversible architectural distortion and functional impairment of the lung parenchyma. - Caused by:
- Most common causes:
- Idiopathic pulmonary fibrosis (IPF, associated with usual interstitial pneumonia [UIP] pattern).
- Connective tissue diseases (e.g., systemic sclerosis, rheumatoid arthritis).
- Other causes include:
- Infectious: Chronic infections such as tuberculosis or histoplasmosis.
- Inflammatory/Immune: Chronic hypersensitivity pneumonitis, sarcoidosis.
- Neoplastic: Post-treatment fibrosis (e.g., radiation-induced fibrosis).
- Mechanical trauma: Chronic aspiration or repetitive mechanical injury.
- Inherited: Genetic interstitial lung diseases, such as surfactant protein mutations.
- Congenital: Rare congenital pulmonary fibrosis.
- Other: Asbestosis, advanced silicosis.
- Most common causes:
- Resulting in:
- Severe architectural distortion of the lung parenchyma.
- Loss of functional alveoli and decreased lung compliance.
- Impaired gas exchange, leading to progressive hypoxia and dyspnea.
- Structural Changes:
- Cystic Airspaces:
- Uniformly sized cystic spaces (3–10 mm) with occasional larger spaces in advanced disease.
- Typically stacked in 2–3 layers, although single-layer honeycombing may occur in early stages.
- Bronchovascular Distortion: Accompanied by traction bronchiectasis.
- Fibrotic Changes: Dense fibrosis replacing normal alveolar structures.
- Cystic Airspaces:
- Pathophysiology:
- Repeated cycles of epithelial injury and repair lead to excessive fibroblast activation.
- Fibroblast proliferation and extracellular matrix deposition destroy normal alveolar structures, forming cystic spaces.
- Mechanical forces, such as tension at the subpleural regions, contribute to the development of honeycombing.
- Honeycomb cysts, despite being lined by bronchiolar epithelium, are isolated from larger airways due to surrounding fibrosis, which severs their functional airway connectivity.
- Pathology:
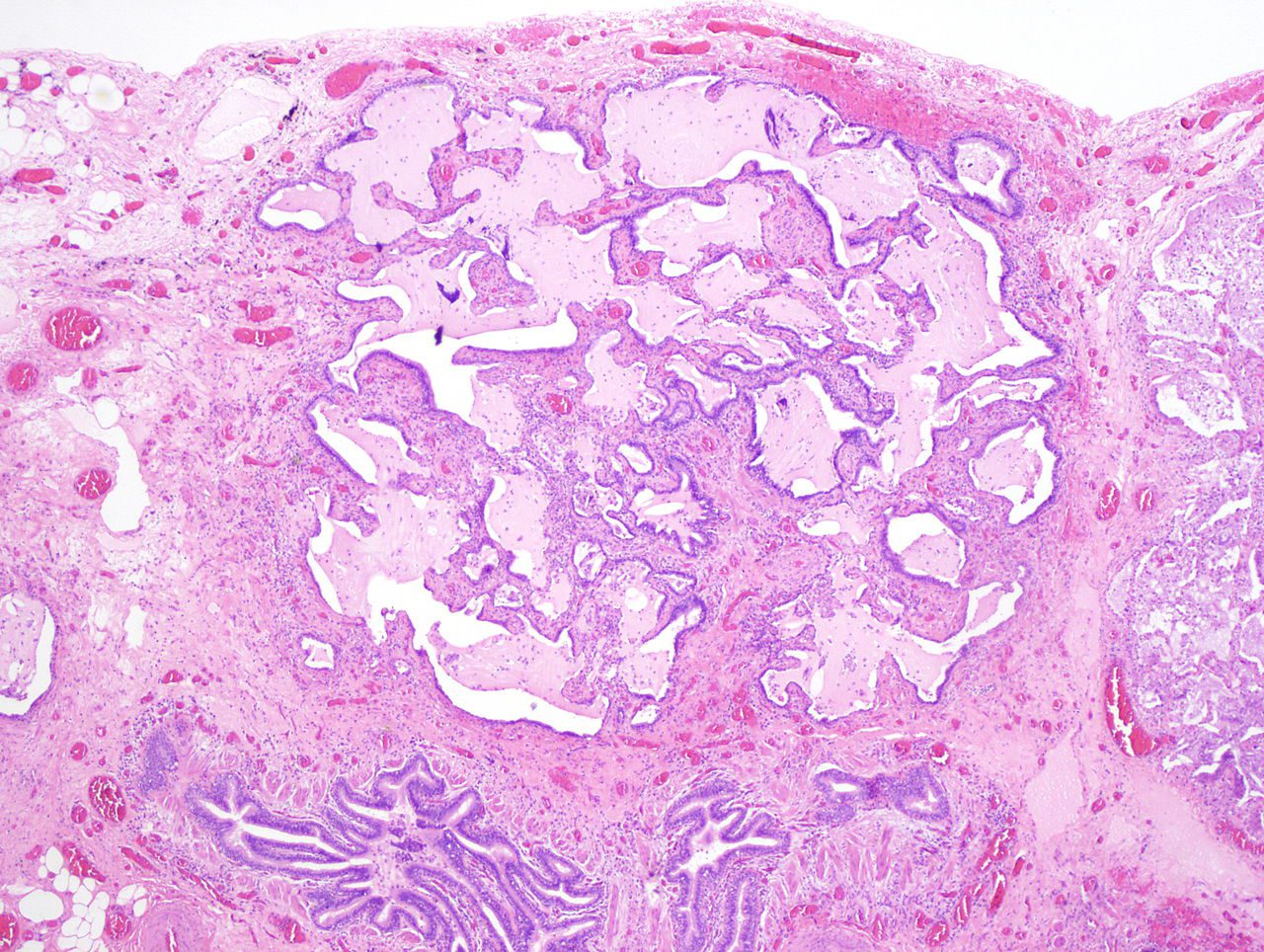
- Hallmark: Cystic spaces lined by bronchiolar epithelium, distinguishing honeycombing from other cystic lung changes.
- Gross: Firm, shrunken lungs with subpleural clusters of cystic spaces.
- Microscopic:
- Bronchiolar epithelium lining cystic spaces.
- Surrounding dense fibrosis indicative of end-stage disease.
- Diagnosis:
- Clinical suspicion based on progressive dyspnea, dry cough, and underlying risk factors.
- Confirmed by imaging and/or biopsy in ambiguous cases.
- Clinical:
- Symptoms: Progressive dyspnea on exertion, dry cough.
- Physical Exam: Inspiratory crackles, clubbing in advanced stages.
- Radiology:
- CXR:
- Findings: Reticular opacities with basal and peripheral predominance.
- Associated Findings: Volume loss, traction bronchiectasis.
- CT (High-Resolution Computed Tomography):
- Parts: Subpleural and basal distribution.
- Size: Uniform cystic spaces measuring 3–10 mm, occasionally larger in advanced disease.
- Shape: Rounded or polygonal cysts clustered in layers.
- Position: Predominantly subpleural, often extending into adjacent parenchyma.
- Character:
- Thick fibrotic walls surrounding cystic spaces.
- Traction bronchiectasis and architectural distortion.
- Lack of significant ground-glass opacities unless overlapping acute processes.
- Time: Chronic finding, representing end-stage disease.
- Associated Findings: Reticulations, traction bronchiectasis, volume loss.
- Distinction from Traction Bronchiolectasis:
- Honeycombing forms clustered cystic spaces with a layered appearance and no continuity with larger airways.
- Traction bronchiolectasis involves irregularly dilated airways that maintain a connection to proximal bronchi.
- Other Relevant Imaging Modalities:
- MRI: Limited utility but may highlight cystic spaces.
- PET-CT: May show metabolic activity in areas of active fibrosis or inflammation.
- Ultrasound: Rarely used but may identify pleural-based abnormalities.
- CXR:
- Pulmonary Function Tests (PFTs):
- Restrictive pattern with decreased total lung capacity (TLC).
- Reduced diffusing capacity of the lungs for carbon monoxide (DLCO).
- Management:
- No definitive cure: Focus on symptom management and slowing disease progression.
- Pharmacologic: Antifibrotic therapies (e.g., pirfenidone, nintedanib for IPF).
- Supportive care: Oxygen therapy, pulmonary rehabilitation.
- Advanced therapies: Lung transplantation in eligible patients.
- Recommendations:
- Early diagnosis and initiation of treatment to slow disease progression.
- Monitor disease progression with serial PFTs and imaging.
- Avoid known exposures (e.g., smoking, occupational dusts).
- Vaccinations to prevent infections in compromised lung function.
- Key Points and Pearls:
- Honeycomb lung is a hallmark feature of end-stage interstitial lung diseases.
- The presence of cystic spaces lined by bronchiolar epithelium is a defining pathological hallmark of honeycombing.
- Honeycombing reflects end-stage fibrosis, with irreversible architectural distortion.
- The subpleural and basal distribution of cystic spaces is highly characteristic on imaging.
- Differentiating honeycombing from traction bronchiolectasis is critical—traction bronchiolectasis involves irregularly dilated airways with continuity to proximal bronchi, while honeycombing forms clustered, stacked layers.
- Uniformly sized cystic spaces (3–10 mm) in 2–3 stacked layers are characteristic, though single layers may occur in early disease.
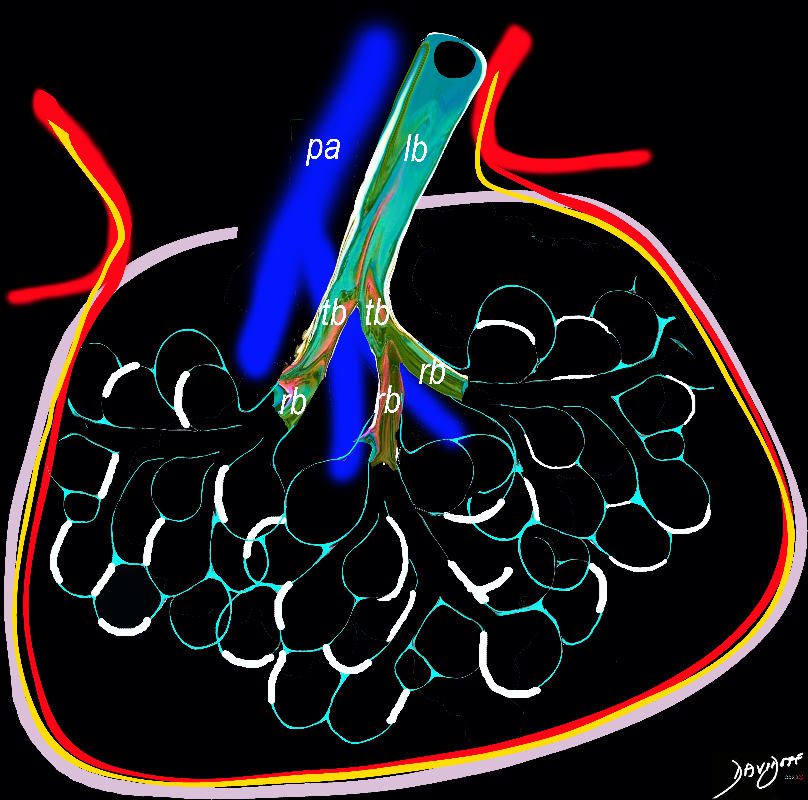
Intralobular, interstial – interalveolar fibrosis (white) between the alveoli
Ashley Davidoff TheCommonVein.net
lungs-0738b
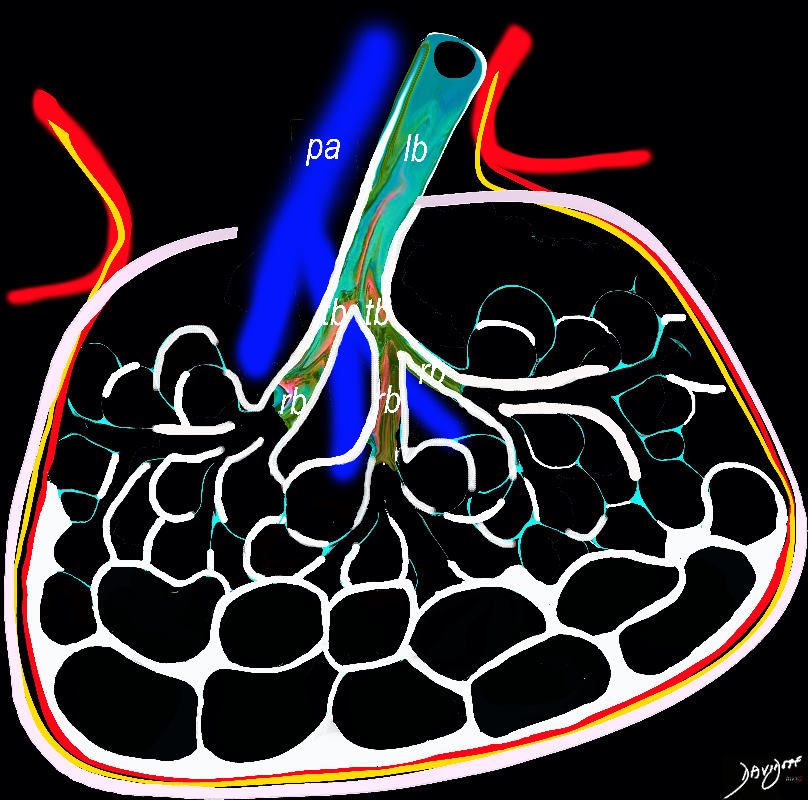
In patients with interstitial lung disease, the inflammatory process and interstitial fibrotic disease progresses and the walls between the alveoli are destroyed causing large subpleural, variably sized, subpleural, thick walled, stacked, cystic spaces . The appearance is reminiscent of a honeycomb and indicates end stage fibrosis
Ashley Davidoff MD thecommonvein.net lungs-0738bh
subpleural regions of the lung and are most severe in the lower lobes and lower portions of all lobes (see the image below).
Ashley Davidoff MD TheCommonVein.net
Source Public Domain
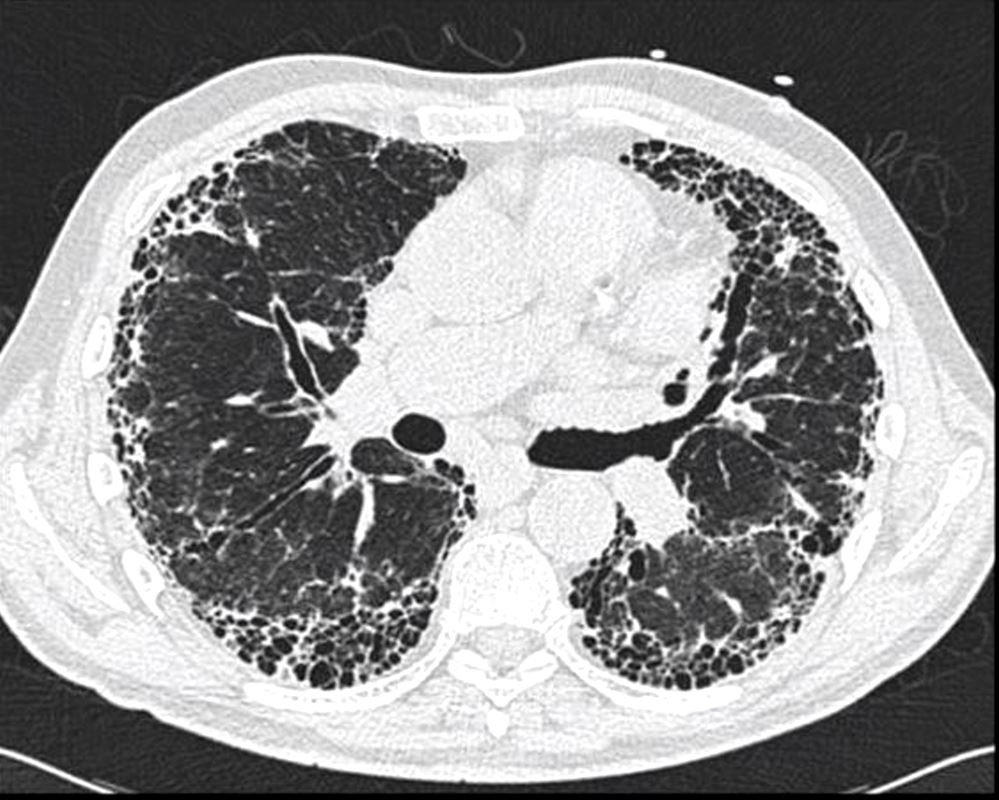
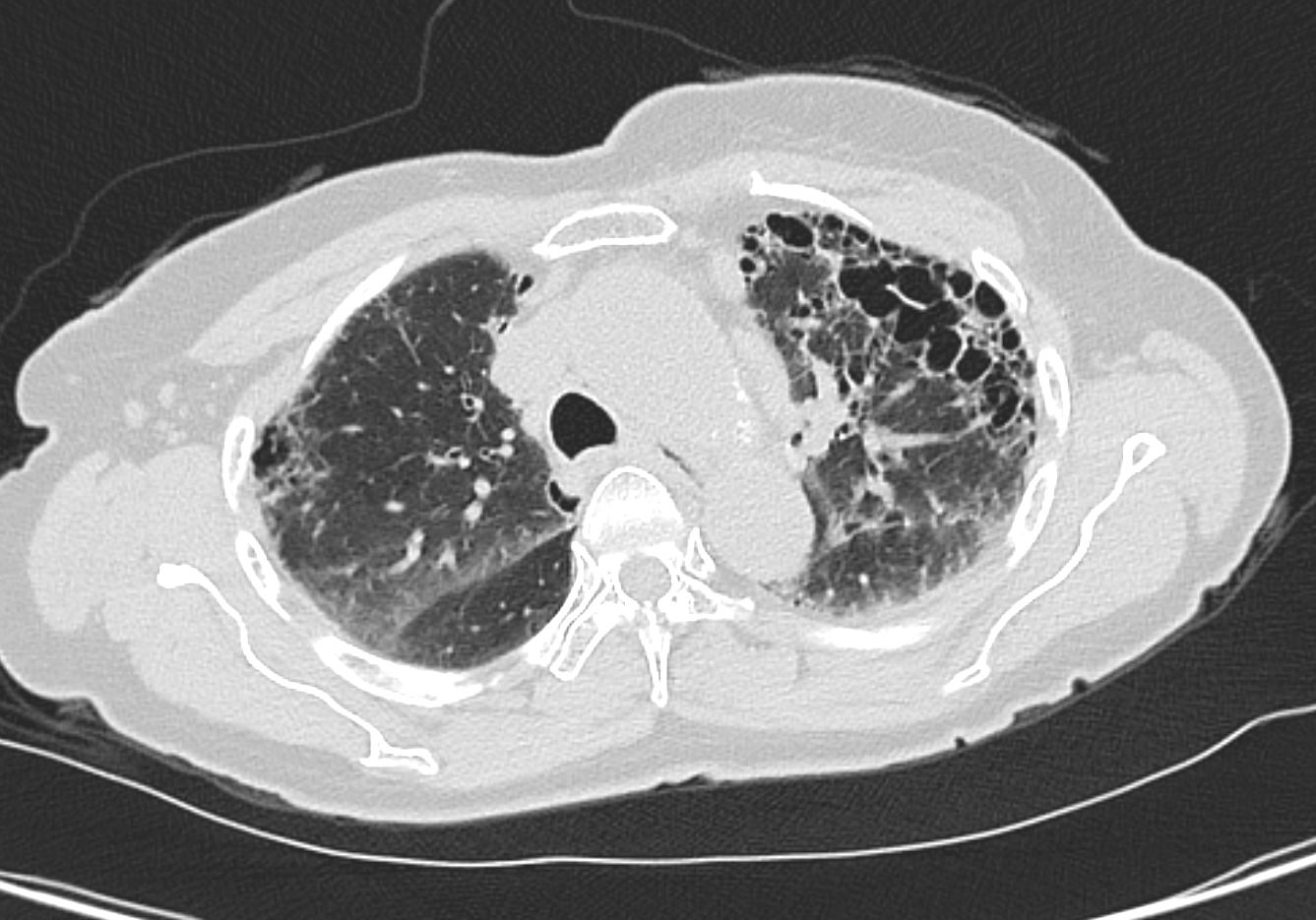
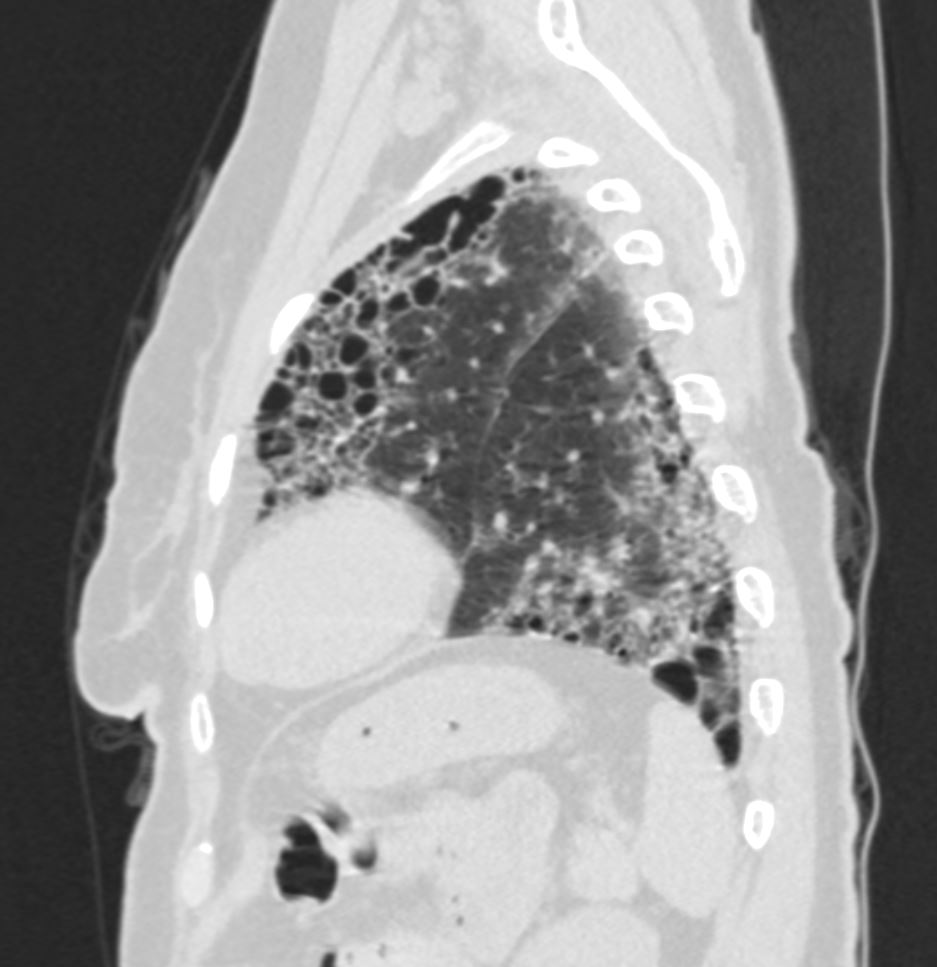
Honeycomb changes in ILD is a feature on CT of interstitial lung disease and is characterised by clusters of stacked cystic air spaces in the periphery and predominantly posteriorly at the bases of the lungs . They likely represent small airways that have been denuded of the their alveoli apparatus. It is most commonly associated with idiopathic pulmonary fibrosis (IPF) and its radiological equivalent UIP – usual interstitial pneumonia.
81f lungs uip 004
Ashley Davidoff
TheCommonVein.net
Ashley Davidoff
TheCommonVein.net
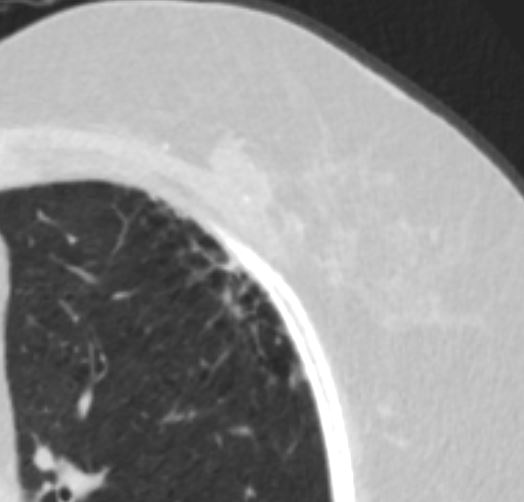
Ashley Davidoff MD TheCommonVein.net honeycomb changes 77-001
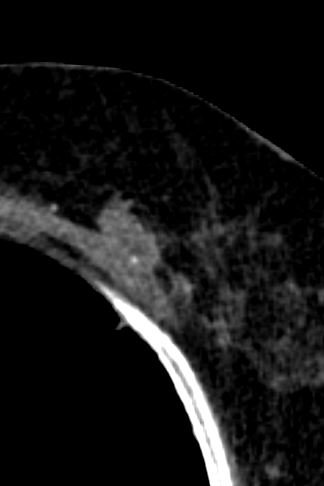
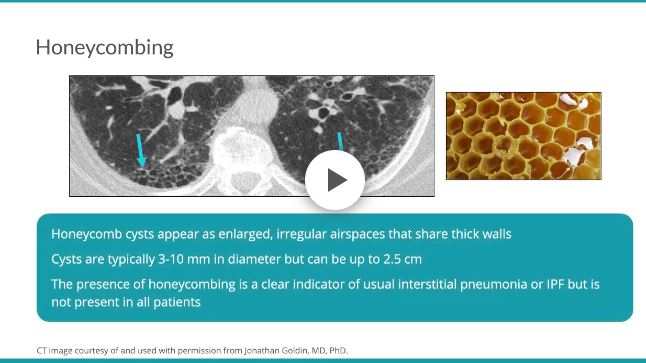
Honeycombing results from the deposition of dense collagen fibers that destroy the characteristic alveolar structure,5 and is typically representative of end-stage lung disease.7 On HRCT, honeycomb cysts appear as enlarged airspaces that are often irregular in size, share thick walls, and are stacked upon one another. The cysts are typically 3-10 mm in diameter but can be as large as 2.5 cm.57 When associated with a pattern of usual interstitial pneumonia typical of idiopathic pulmonary fibrosis, honeycombing typically has a peripheral, basal, and subpleural distribution.45 The percentage of patients with IPF with honeycombing on HRCT varies in the literature, but it is estimated to be observed in one-third to two-thirds of patients.89
Radiology Rounds
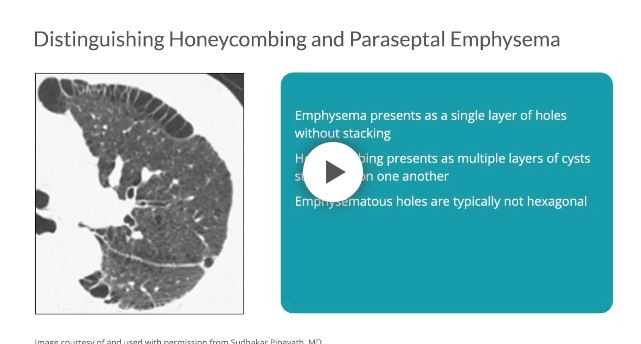
Distinguishing between honeycombing and paraseptal emphysema may be difficult, especially when coexisting on a single scan. As compared with honeycombing, which may present as multiple layers of cysts stacked upon one another, emphysema presents as a single layer of holes without stacking.7 Furthermore, emphysematous holes are typically not hexagonal; therefore, the shape of the cysts and their propensity to stack can help to distinguish one from the other.7
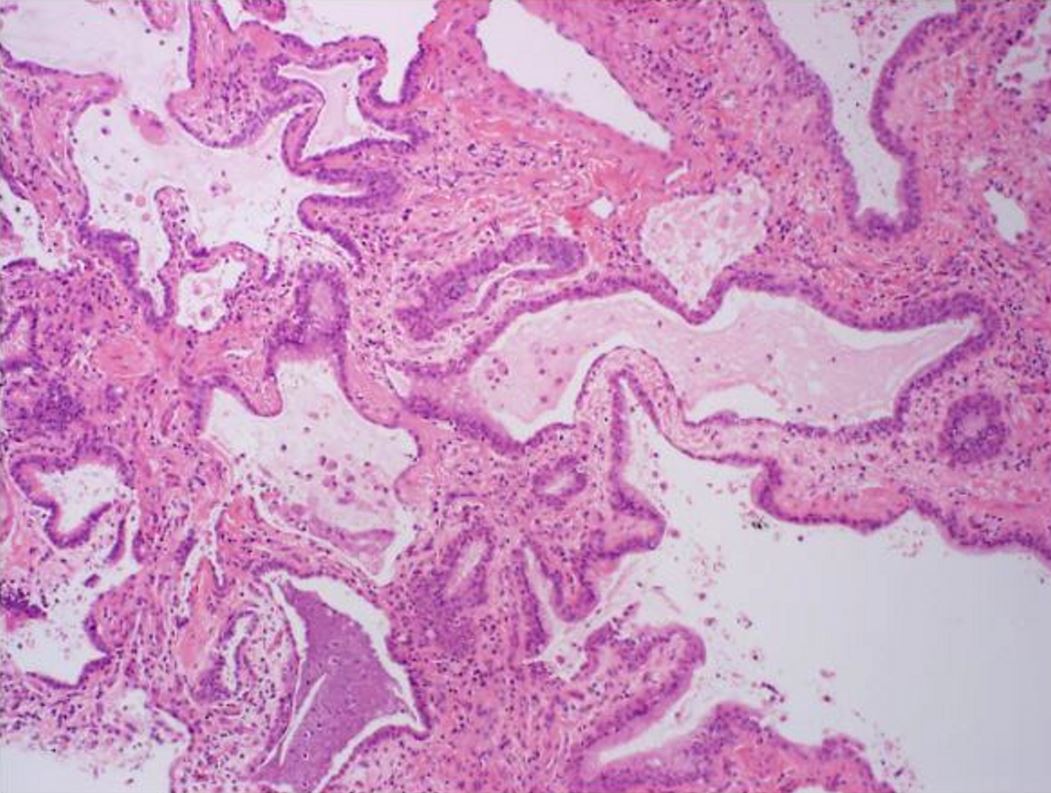
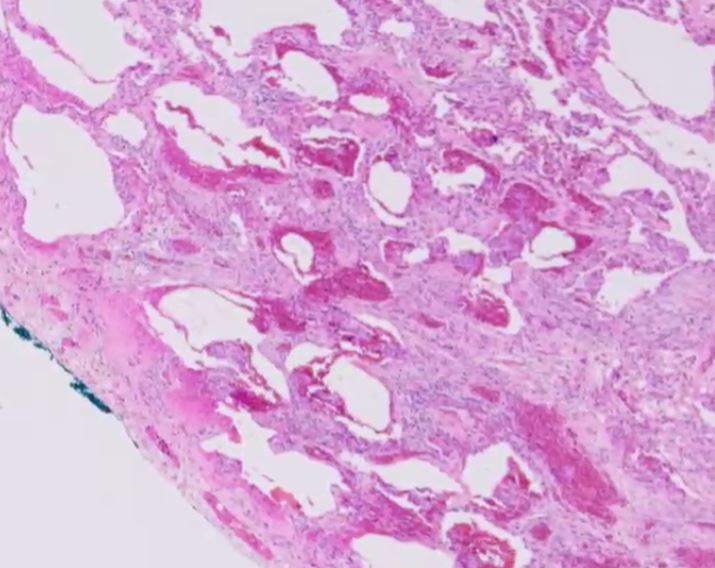
Histologic specimen shows dilated air spaces lined by bronchiolar type epithelium and surrounding fibrosis consistent with microscopic honeycomb change. Courtesy Medscape

Typical histological finding of spatial heterogeneity in usual interstitial pneumonia (UIP) with an abrupt transition from honeycomb change (left) to normal lung (right) using trichrome stain, X40)
Courtesy Medscape eMedicine

Axial CT through the base of the lungs shows honeycombing change sin the posterior segment of the left lower lobe characterized by the stacking of cystic spaces.
Ashley Davidoff MD
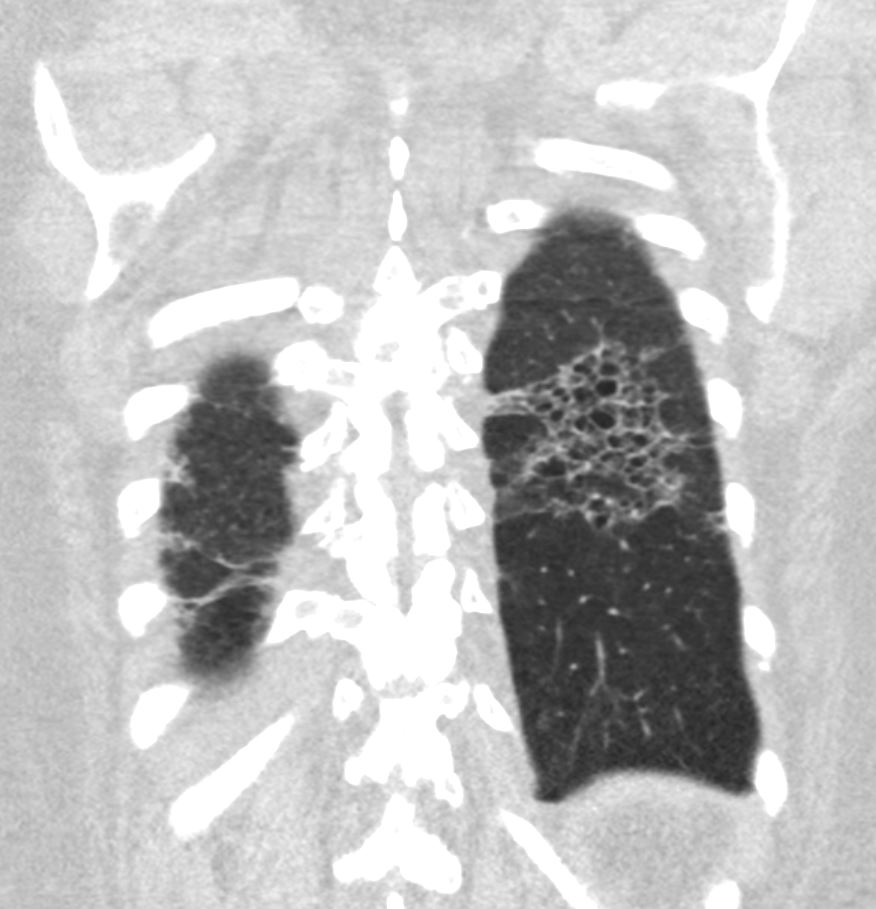
Coronal reconstruction CT through the posterior portion of the lungs shows honeycombing change sin the posterior segment of the left lower lobe characterized by the stacking of cystic spaces.
Ashley Davidoff MD
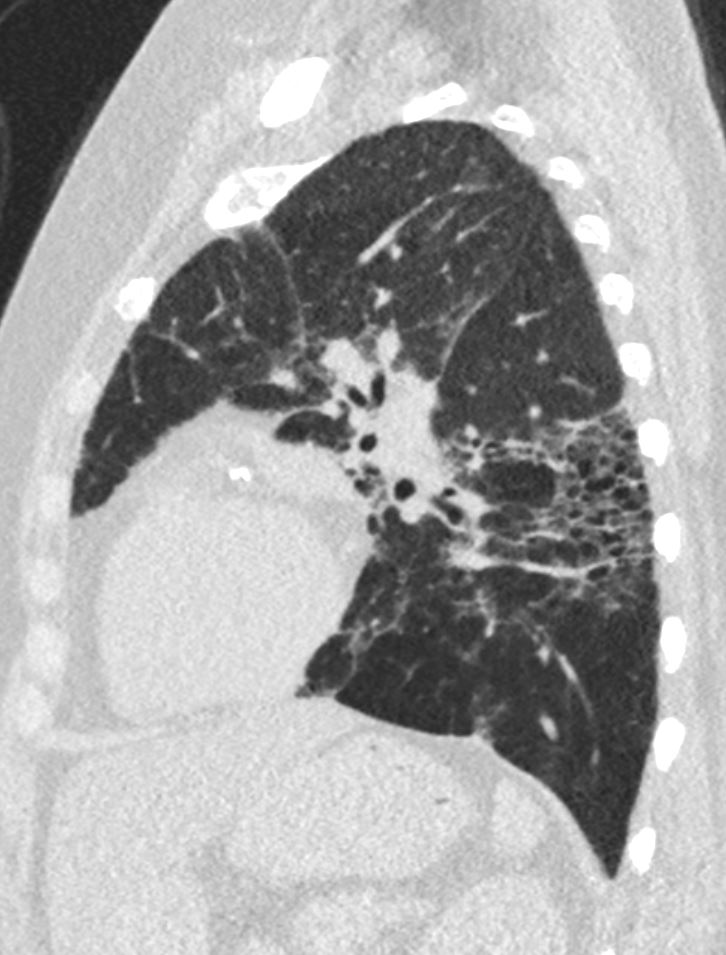
Sagital reconstruction CT through the posterior portion of the lungs shows honeycombing change sin the posterior segment of the left lower lobe characterized by the stacking of cystic spaces.
Ashley Davidoff MD
“In UIP, the fibrotic process produces
patchy but diffuse changes in end-stage lung disease. Endstage lung disease, as defined morphologically, represents the
culmination of a ‘‘dying-back’’ process of acini within the
secondary pulmonary lobule. One may conceptualize this
with respect to lung embryogenesis. The lung in utero
develops from the foregut and undergoes pseudoglandular,
canalicular, and alveolated stages, which roughly correspond with the trimesters of pregnancy, respectively. The
mature lung is capable of gas-exchange because of
alveolation. Usual interstitial pneumonitis, in an admittedly
crude manner, reverses this sequence, so that the alveolated
lung is deconstructed to a set of ‘‘glandular’’ airways
surrounded by fibrosis.
Lung: Time for a Change—Kradin
What is the ‘‘end-stage’’ of the acinus? When the
alveolated structures are lost, a distorted ectatic terminal
bronchiole, lined by bronchiolar epithelium, is left, which is
why the changes of UIP are dominantly subpleural or
periseptal, because this is where distal lung acini terminate.
Accordingly, end-stage lung disease should show no
intervening healthy alveolated structures between the
ectatic bronchiole and the subjacent visceral pleura or septa.
The HRCT appearance of end-stage lung should include
microcysts associated with scar and no evident distal
alveolated lung parenchyma.
Currently, the early radiographic diagnosis of UIP is
hampered by 2 factors: the requirement for identifying
honeycomb lung, the other, confusion with traction bronchiolectasis. Honeycomb lung is reminiscent of the appearance
of the honeycomb of a beehive. This reflects the stacking of
ectatic bronchioles in which no alveolated lung intervenes. It
is not, however, a biological term and, therefore, lacks the
sensitivity of the pathologist’s microscopic examination
where foci of end-stage lung disease can be identified with
confidence before the stacking of ectatic bronchioles has
developed. In my experience, cystic bronchiolectasis can be
seen in computed tomography scans without honeycombing,
and many of these cases will prove to have UIP on biopsy. A
careful examination of the subpleural lung should show no
intervening zone of aerated lung between the microcysts and
the visceral pleura. The recent introduction of dual-energy
HRCT scanning may increase the sensitivity for detecting
alveolated structures, allowing for an earlier, noninvasive
diagnosis of UIP”
I recommend that the phrase intralobular fibrous obliteration with microcysts be adopted by both radiologists and
pathologists to reflect the bronchiolectasis and peripheral
scarring of the end-stage lung in UIP. Even a few subpleural
microcysts in the periphery of a diffusely fibrotic lung may represent early UIP. The current binary reading, based on
the presence or absence of honeycomb lung, potentially
renders a disservice to clinicians who must make critical
diagnostic and therapeutic decisions. To avoid reducing the
specificity of computed tomography scanning in the
diagnosis of UIP, the phrase intralobular fibrous obliteration
with microcysts may require modifiers. Three examples of
suggested HRCT diagnoses are given below:
1. ‘‘Diffuse patchy interstitial fibrosis with subpleural
intralobular fibrous obliteration showing multiple microcysts. This appearance is diagnostic for UIP.’’
2. ‘‘Diffuse interstitial fibrosis with occasional subpleural
intralobular cystic spaces. This appearance favors early
UIP, but biopsy confirmation may be indicated.’’
3. ‘‘Diffuse interstitial fibrosis with cystic airways with a zone
of alveolated lung identified subjacent to the visceral
pleura. The findings are most consistent with nonspecific
interstitial pneumonitis and traction bronchiectasis. Clinical and pathologic correlation may be required.’’
The phrase intralobular fibrous obliteration with cystic
changes is accurate; honeycomb lung, although commonplace
and widely adopted, is difficult to define and meaningless
biologically. It is also insensitive to early or milder disease.
In addition, the term honeycomb lung is not necessarily
helpful for pathologists who may think they know what it is. In
classic cases of UIP, the cystic bronchioles should be lined by
respiratory ciliated epithelium, but in a subset of cases, the
cysts of diffusely fibrotic lungs are instead lined by cuboidal
epithelium. These examples of honeycomb lung show distinct
epithelial expression of FoxF1 and SSH proteins, gene
products that have a role in lung development.7 I suggest that
pathologists pay attention not only to whether there is patchy
diffuse scarring and microcystic, honeycomb’’ changes in
diagnosing UIP but also to the nature of the epithelium lining
the microcysts because this may reflect distinct pathways of
structural simplification and lung remodeling with pathogenetic and clinical significance. In my own practice of
pulmonary pathology, I limit a definitive diagnosis of UIP to
cases where cystic bronchiolectasis with respiratory epithelium
is identified. Attempting to cubbyhole diagnoses likely will not
advance our knowledge of these diseases. Therefore, adhering
to strict criteria for what represents the diagnosis of UIP may
prove to be an important advance for both the diagnosis and
the ultimate understanding and treatment of this disorder”
Reprints: Richard L. Kradin, MD, Department of Pathology,
Massachusetts General Hospital, 55 Fruit St, Warren 253, Boston,
MA 02114-2696 (e-mail: rkradin@partners.org).
1398 Arch Pathol Lab Med—Vol 139, November 2015 Honeycomb