- LIP is a rare interstitial lung disease characterized by the diffuse proliferation of lymphoid tissue within the lung interstitium.
- In lymphocytic interstitial pneumonia (LIP), there is an inflammatory component as well as infiltrative features.
-
Etymology
- Derived from “lymphocytic,” referring to lymphocytes, and “interstitial pneumonia,” indicating inflammation primarily affecting the lung interstitium.
AKA and abbreviation
- Lymphocytic interstitial pneumonia (LIP).
- What is it:
- LIP is a rare, benign lymphoproliferative disorder characterized by diffuse lymphocytic infiltration of the pulmonary interstitium, alveolar septa, and associated lymphatic structures.
- is a rare form of interstitial lung disease (ILD) characterized by:
- Diffuse or patchy lymphocytic infiltration of the
- airways
- interstitium and
- alveolar walls
- Associated with
- It often occurs in association with autoimmune diseases, immunodeficiency syndromes, or chronic antigenic stimulation.
- hyperplasia of lymphoid tissue in the lungs.
- Diffuse or patchy lymphocytic infiltration of the
- LIP can occur as an
- idiopathic condition or
- secondary to other systemic diseases.
- is a rare form of interstitial lung disease (ILD) characterized by:
-
Characterized by
- Histological findings:
- Dense interstitial infiltration of mature lymphocytes, plasma cells, and histiocytes.
- Prominent lymphoid aggregates with or without germinal centers in the interstitium.
- Sparing of the airways in most cases.
- Imaging features:
- Diffuse or patchy ground-glass opacities.
- Perilymphatic or randomly distributed thin-walled cysts.
- Reticulonodular opacities and septal thickening.
- Upper and lower lobe involvement with subpleural predominance.
- Associated findings:
- Mediastinal or hilar lymphadenopathy.
- Pleural effusions (rare).
Anatomical Context
- Primarily affects the pulmonary interstitium, including:
- Alveolar walls.
- Peribronchovascular lymphatic structures.
- Subpleural and interlobular septal regions.
Caused by
Most common cause: Autoimmune diseases.
Other causes include:
- Autoimmune diseases:
- Sjögren syndrome (most common).
- Systemic lupus erythematosus (SLE).
- Rheumatoid arthritis.
- Immunodeficiency syndromes:
- Common variable immunodeficiency (CVID).
- HIV/AIDS (especially in children).
- Virus-associated:
- Epstein–Barr virus (EBV).
- Human immunodeficiency virus (HIV).
- Other:
- Idiopathic cases.
- Post-transplant immune reconstitution.
Resulting in
- Chronic lymphocytic infiltration of the interstitium.
- Diffuse pulmonary inflammation, potential fibrosis, and cystic changes.
- Impaired gas exchange and restrictive lung physiology.
Structural changes
- Inflammation: Diffuse lymphocytic infiltration in the interstitial tissue.
- Cysts: Thin-walled cysts due to peribronchial lymphoid tissue hyperplasia and alveolar destruction.
- Fibrosis: In advanced cases, interstitial fibrosis may develop.
Pathophysiology
- Chronic antigenic stimulation triggers lymphocytic proliferation within the pulmonary interstitium.
- Progressive inflammation leads to diffuse interstitial involvement and the formation of lymphoid follicles.
- Cystic changes occur due to obstruction of bronchioles by peribronchial lymphoid aggregates.
Pathology
- Interstitial expansion by lymphocytic infiltrates, predominantly CD4+ and CD8+ T cells.
- Follicular hyperplasia with germinal centers.
- Minimal to no granulomatous inflammation.
Diagnosis
- Clinical correlation: Symptoms such as cough, dyspnea, and systemic features like fever or weight loss.
- Imaging:
- HRCT: Diffuse ground-glass opacities, reticulonodular patterns, and cysts.
- CXR: Reticulonodular opacities; less specific than CT.
- Biopsy: Definitive diagnosis requires histopathological confirmation.
Clinical
- Symptoms:
- Insidious onset of dyspnea and non-productive cough.
- Systemic symptoms: Fatigue, weight loss, low-grade fever.
- Signs:
- Inspiratory crackles (in advanced cases).
- Extrapulmonary manifestations of associated autoimmune diseases (e.g., xerostomia in Sjögren syndrome).
Radiology Detail
CXR:
- Findings: Reticulonodular opacities with lower lobe predominance.
CT:
- Parts: Alveolar walls, peribronchovascular interstitium, and subpleural regions.
- Size: Cysts typically measure 1-3 cm.
- Shape: Round, thin-walled cysts; diffuse ground-glass opacities and reticular changes.
- Position: Predominantly basal and subpleural distribution.
- Character:
- Mixed reticular and ground-glass patterns.
- Associated nodules and cysts.
Other relevant Imaging Modalities:
- MRI: Limited utility for LIP.
- PET-CT: May show mild metabolic activity in lymphoid aggregates.
Pulmonary function tests (PFTs)
- Restrictive pattern with reduced lung volumes (e.g., FVC, TLC).
- Impaired diffusion capacity (DLCO).
Recommendations
- Imaging follow-up: Periodic HRCT to monitor disease progression or response to treatment.
- Biopsy: Required for definitive diagnosis in ambiguous cases.
- Treatment:
- Treat underlying autoimmune or immunodeficiency conditions.
- Corticosteroids or immunosuppressants for progressive disease.
- Antiretroviral therapy in HIV-associated LIP.
Key Points and Pearls
- LIP is strongly associated with autoimmune diseases (e.g., Sjögren syndrome) and immunodeficiency syndromes (e.g., CVID).
- Characteristic imaging findings include diffuse ground-glass opacities, cysts, and reticulonodular patterns.
- Diagnosis requires integration of clinical, radiologic, and histopathologic findings.
- Untreated, LIP may progress to interstitial fibrosis or transform into lymphoma, particularly in the setting of immunodeficiency.
- Histological findings:
Inflammatory Component in the
Interalveolar Septa
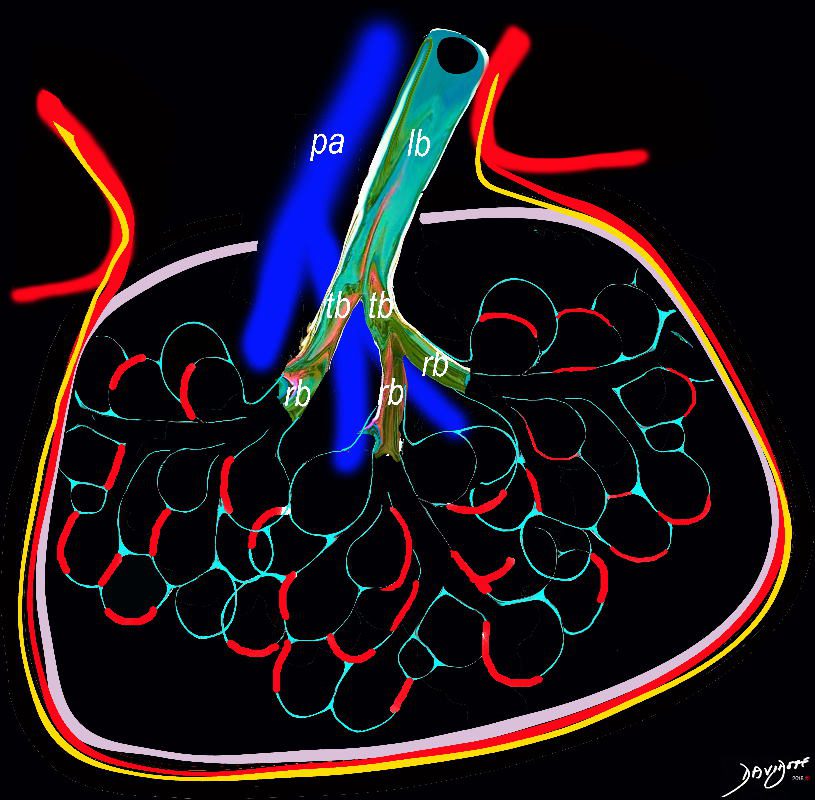
Ashley Davidoff TheCommonVein.net lungs-0736a01
Infiltrative Component of
Lymphocytes Plasma Cell And Histiocytes
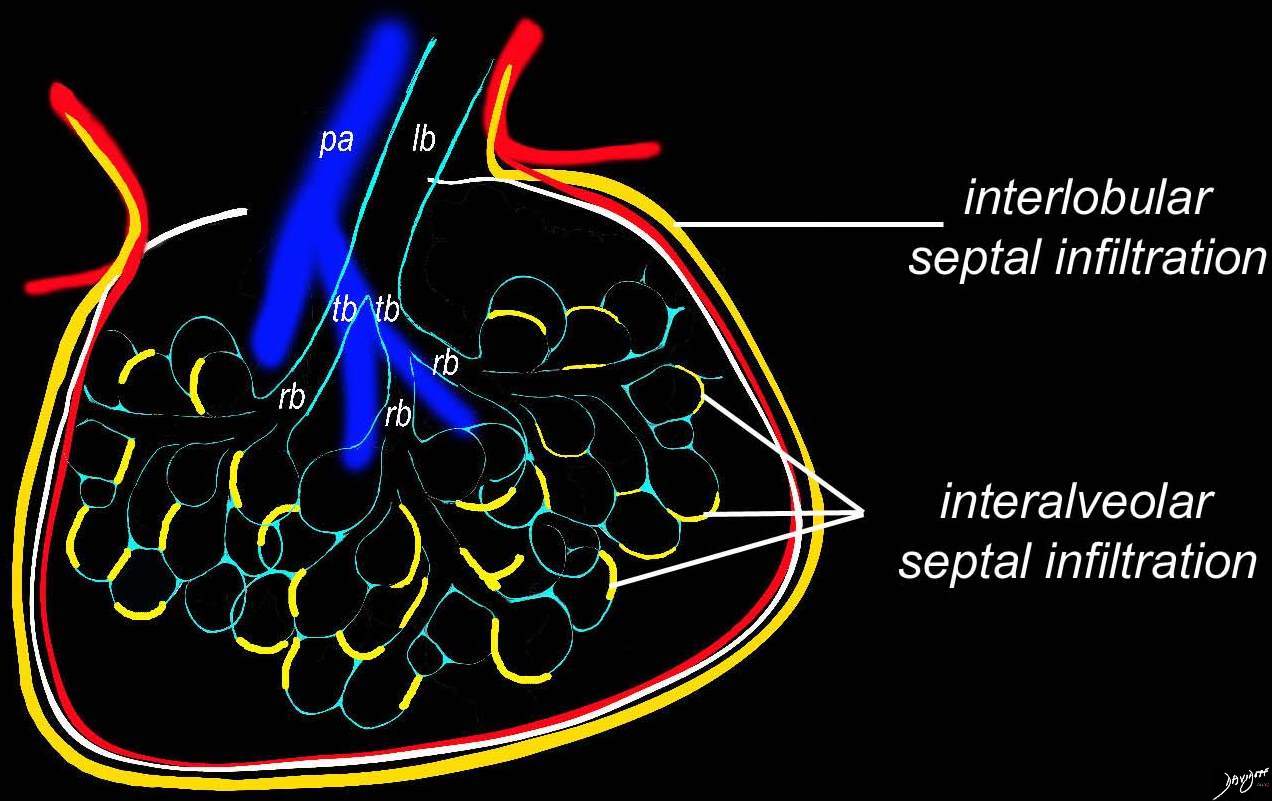
Artistic rendition that shows the position of the lymphatic involvement of cellular infiltration along the interalveolar septa and interlobular Septa dominantly affecting the lower lobes
Ashley Davidoff MD TheCommonVein.net 32645a06L01 lymphatic
Key Pathological Features
- Inflammatory Component:
- LIP is primarily driven by a polyclonal lymphocytic infiltration of the interstitium.
- This inflammation predominantly consists of T-cells, with smaller numbers of B-cells and plasma cells.
- The inflammation may also extend to involve alveolar walls, airways, and blood vessels.
- Infiltrative Features:
- Reactive lymphoid hyperplasia is a hallmark, with scattered lymphoid aggregates sometimes forming germinal centers.
- These lymphoid infiltrates can involve peribronchovascular areas, interlobular septa, and subpleural regions.
- Associated Findings:
- Occasionally, there is infiltration of plasma cells that may produce immunoglobulin, potentially leading to the presence of monoclonal protein production in rare cases (e.g., in the context of Sjögren’s syndrome).
- The alveolar spaces may show collections of macrophages and lymphocytes.
Imaging Correlation
On HRCT, LIP typically shows:
- Diffuse or patchy ground-glass opacities (inflammatory component).
- Thin-walled cysts scattered in a random distribution.
- Reticular or nodular opacities, particularly in the peribronchovascular or lower lung zones.
- In severe or chronic cases, fibrosis may develop.
Clinical Significance
The inflammatory nature of LIP is critical because it means the disease can respond to immunosuppressive therapy (e.g., corticosteroids or other immunomodulatory agents). However, progression to fibrosis or transformation into lymphoid malignancy (e.g., lymphoma) is a potential concern.
In summary, LIP includes both an inflammatory and an infiltrative component, with the inflammatory aspect being central to its pathogenesis and clinical management.
- Lower and midlung predominance
- infiltration of lymphocytes and plasma cells into the
- perilymphatic
- bronchovascular bundles
- venules
- interstitium.
- perilymphatic
Lymphocytic Infiltration Along the
Bronchovascular Bundles in the Mid and
Lower Lung Fields
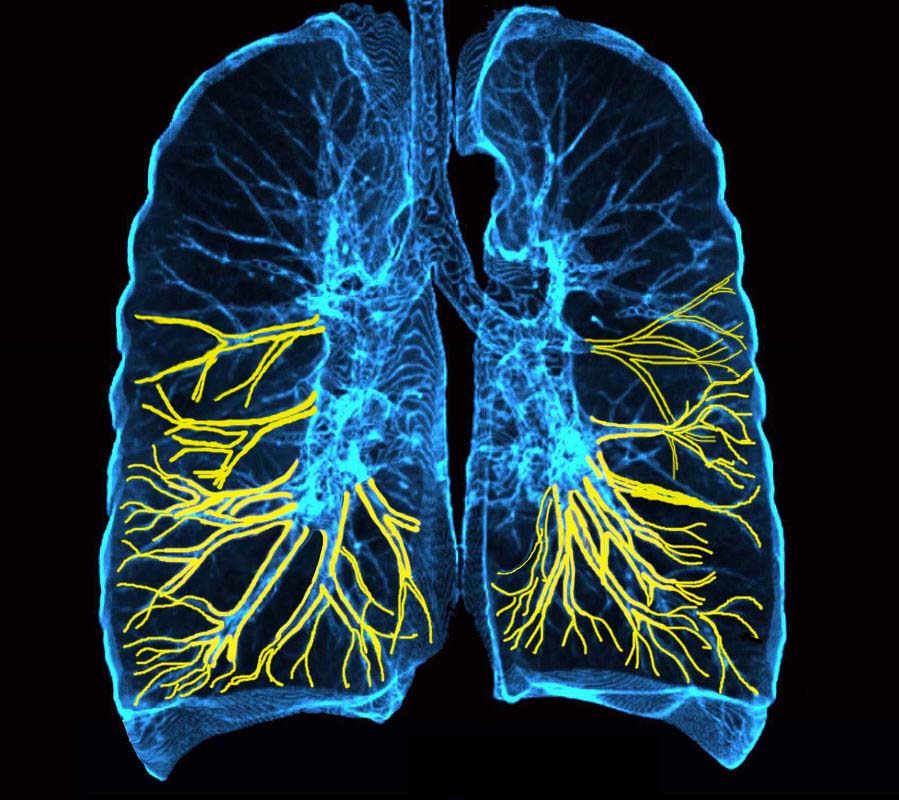
3D reconstruction of a CT scan shows the position of the lymphatic involvement of cellular infiltration along the bronchovascular bundle to the lower lobes
Ashley Davidoff MD TheCommonVein.net
32645a06b01lymphatics
Lymphocytic Infiltration Along the
Lymphatics of the
Interalveolar and
Interlobular Septa
Artistic rendition that shows the position of the lymphatic involvement of cellular infiltration along the interalveolar septa and interlobular Septa dominantly affecting the lower lobes
Ashley Davidoff MD TheCommonVein.net 32645a06L01 lymphatic
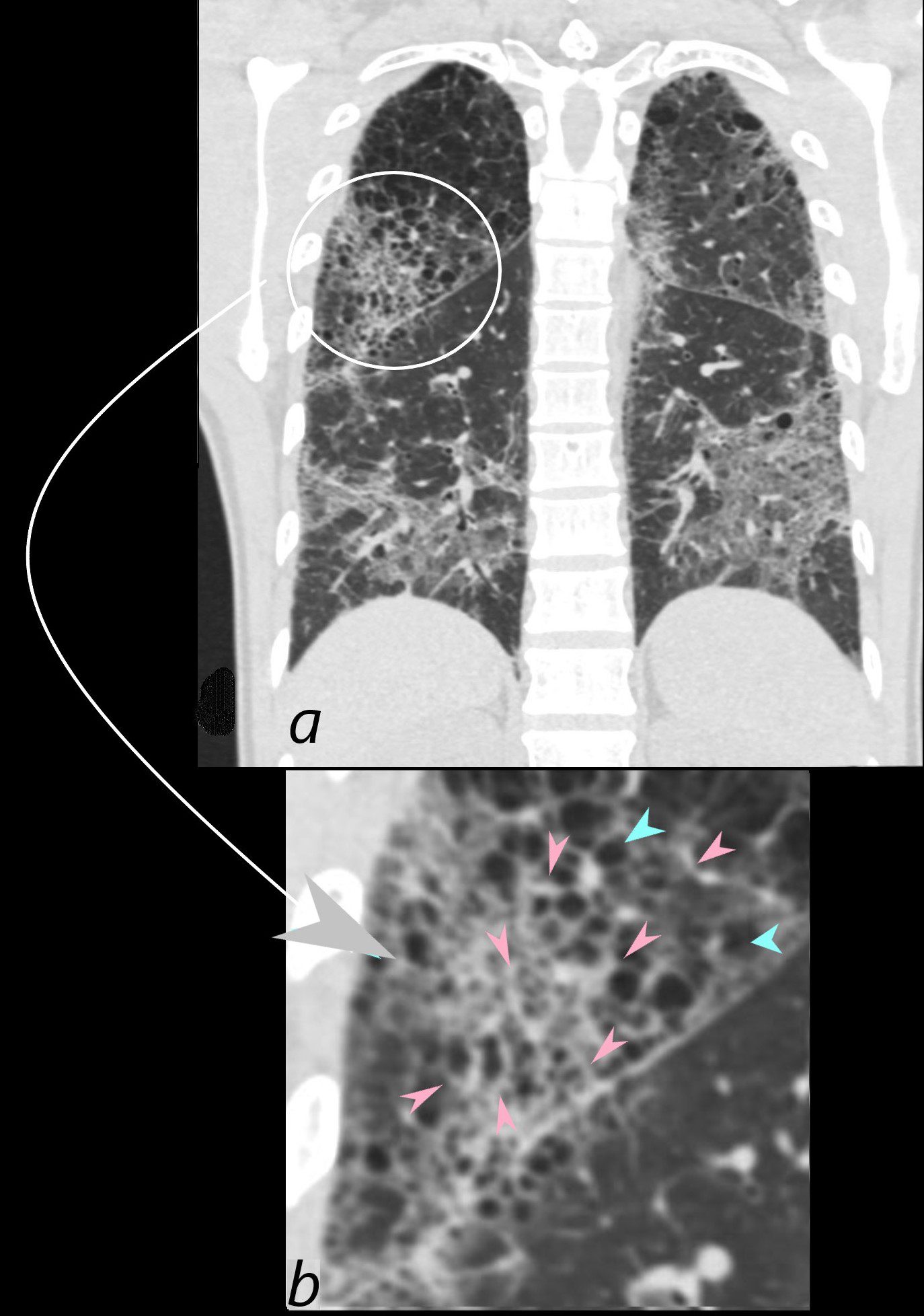
CT scan in coronal projection (a) shows multifocal regions of ground glass opacity in both the lower and upper lobes, associated with mild centrilobular emphysema. The right upper lobe changes are magnified in b and show a reticular pattern as a result of interlobular septal thickening (pink arrowheads in b) The cystic changes seen above (teal blue arrowheads) are part of an underlying emphysematous process. The ground glass changes are as a result of the combination of interalveolar septal and interlobular septal changes resulting in an overall intermediate density of the affected lung and exemplified in the left lower lobe (a)
Ashley Davidoff MD The CommonVein.net 139260c01L 28Lu
Infiltration of Lymphocytes, Plasma Cells, and Histiocytes into the
Walls of the Bronchioles and Small Airways, Obstruction,
Necrosis and
Cyst Formation
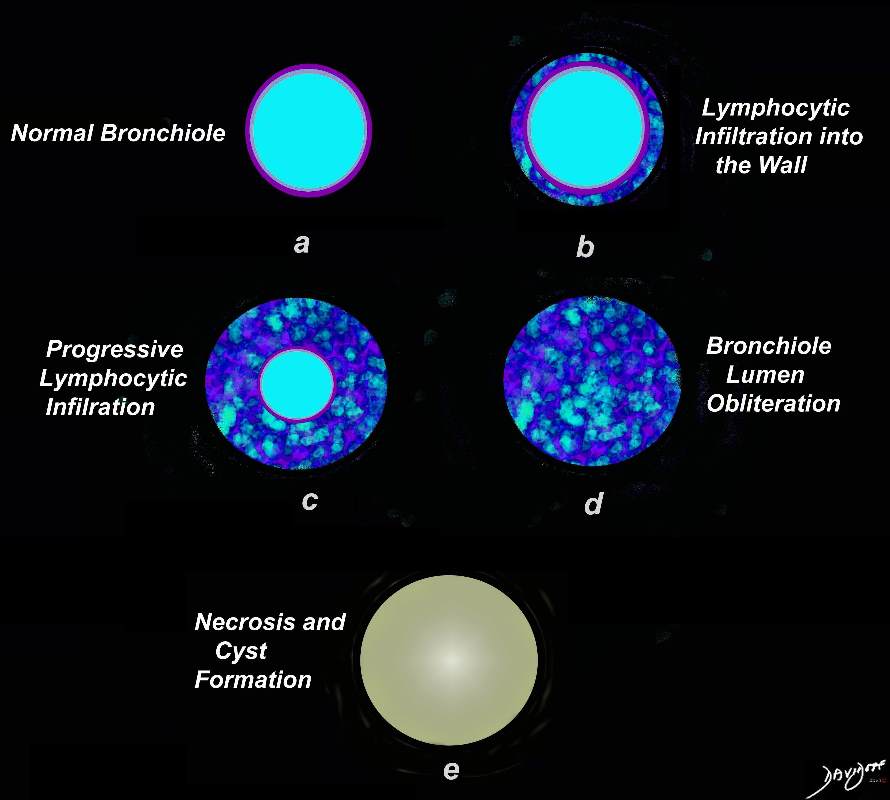
Artistic rendering shows a normal bronchiole (a) with early infiltration into the wall of the airway, (b) progressing to luminal compromise (c) and subsequent obliteration of the lumen (d). Radiologically this manifests as centrilobular nodules. Subsequently there is necrosis and a thin walled cyst remains (e)
Ashley Davidoff MD TheCommonVein.net 139264.lungs
Mid and Lower Lung Field
Thin Walled Cysts
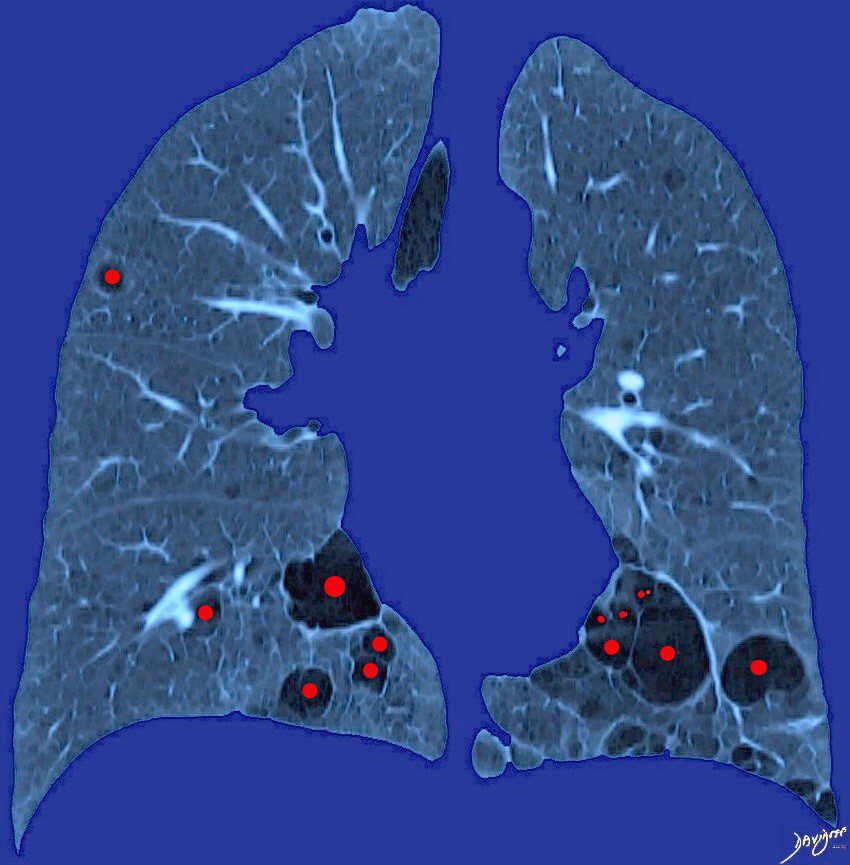
58 year old female with known Sjogren’s syndrome presents for CT evaluation of the chest. Artistic rendition of her CT in the coronal plane shows multiple thin walled cysts in close association with bronchovascular bundles involving the lower and mid lung fields. Biopsy confirmed the associated presence of amyloidosis and LIP (lymphocytic interstitial pneumonia)
Thin Walled Cysts in
Close Association with Bronchovascular Bundles

The cysts of LIP (lymphocytic interstitial pneumonitis) are characteristically associated with the bronchovascular bundle (arteriole dark blue, bronchiole teal).
Ashley Davidoff MD TheCommonVein.net 139265.lungs
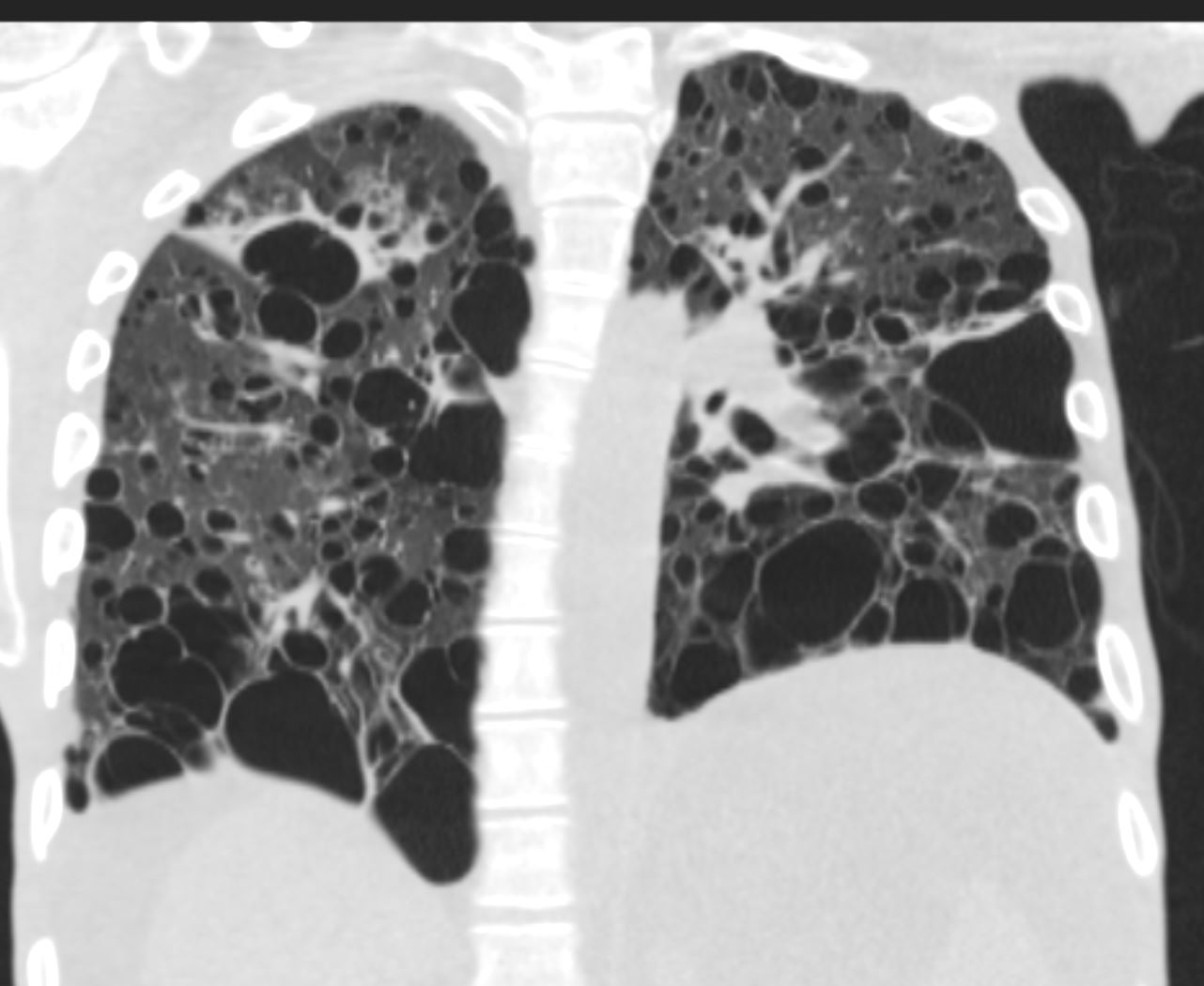
27 year old male with a history of perinatal HIV and lymphocytic interstitial pneumonitis
CT in the coronal plane confirms the presence of diffuse cystic changes with the largest cysts at the lung bases but also involving the mid and upper lung fields .Focal band of ground glass opacity noted in the right upper lobe
Ashley Davidoff MD TheCommonVein.net 139275 017Lu
Nodules
Ill Defined Centrilobular and Subpleural Nodules
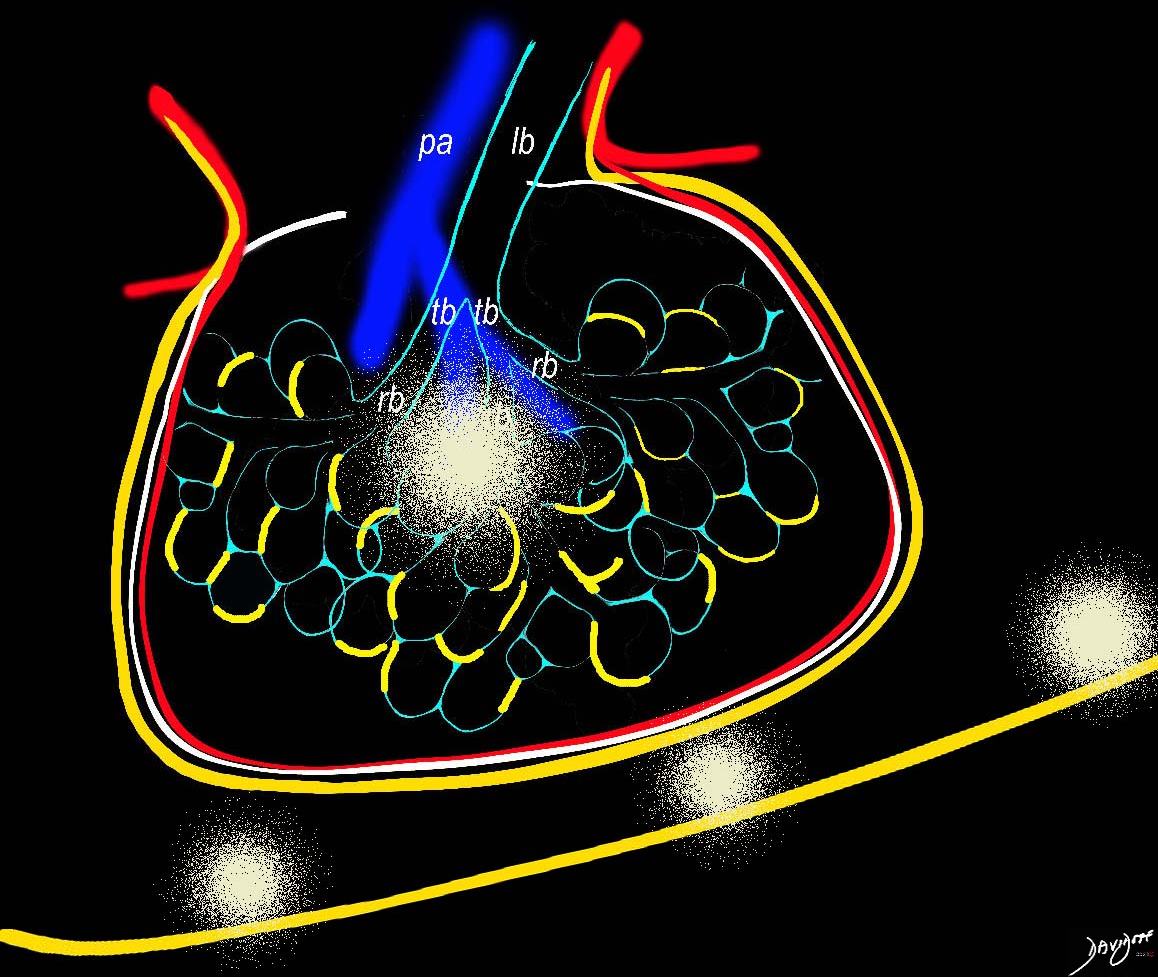
The infiltration of lymphocytes, histiocytes and plasma cells along the lymphatics of the lungs, can sometimes result in ill defined nodules that may be centrilobular or subpleural in location
Ashley Davidoff MD TheCommonVein.net
32645a06L
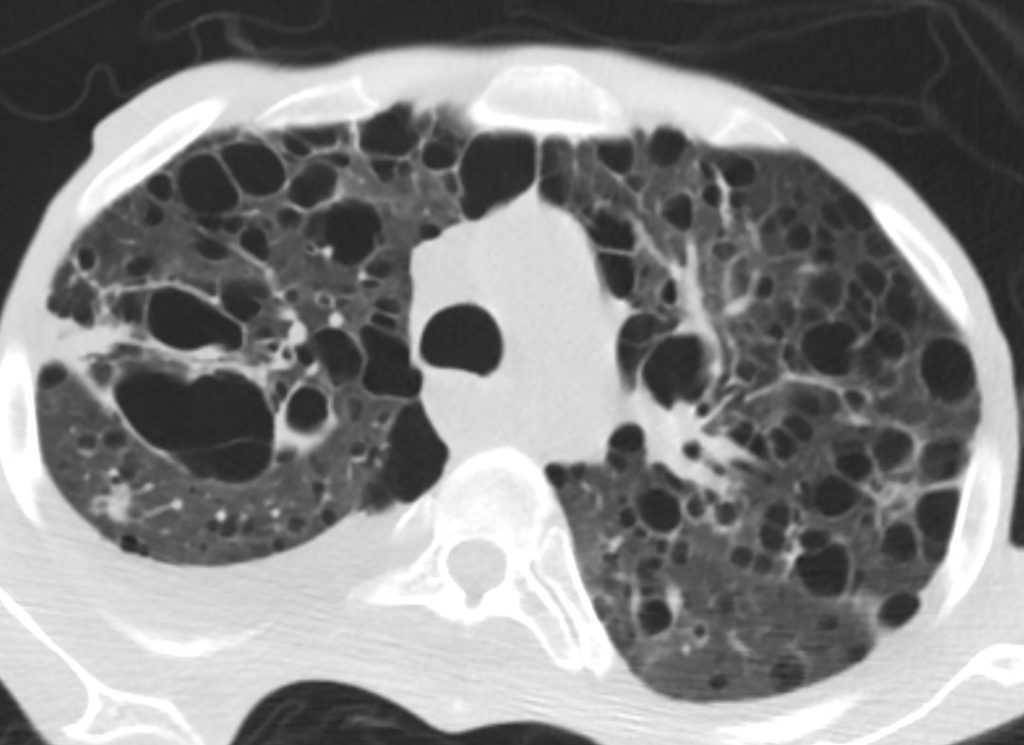
27 year old male with a history of perinatal HIV and lymphocytic interstitial pneumonitis
CT in the axial plane confirms the presence of diffuse thin walled cystic changes . Focal band of ground glass opacity noted in the right upper lobe
A mixed density 8mm nodule is noted in the posterior segment of the right upper lobe. There is a new small to moderate right sided pleural effusion.
Ashley Davidoff MD TheCommonVein.net 139276 017Lu
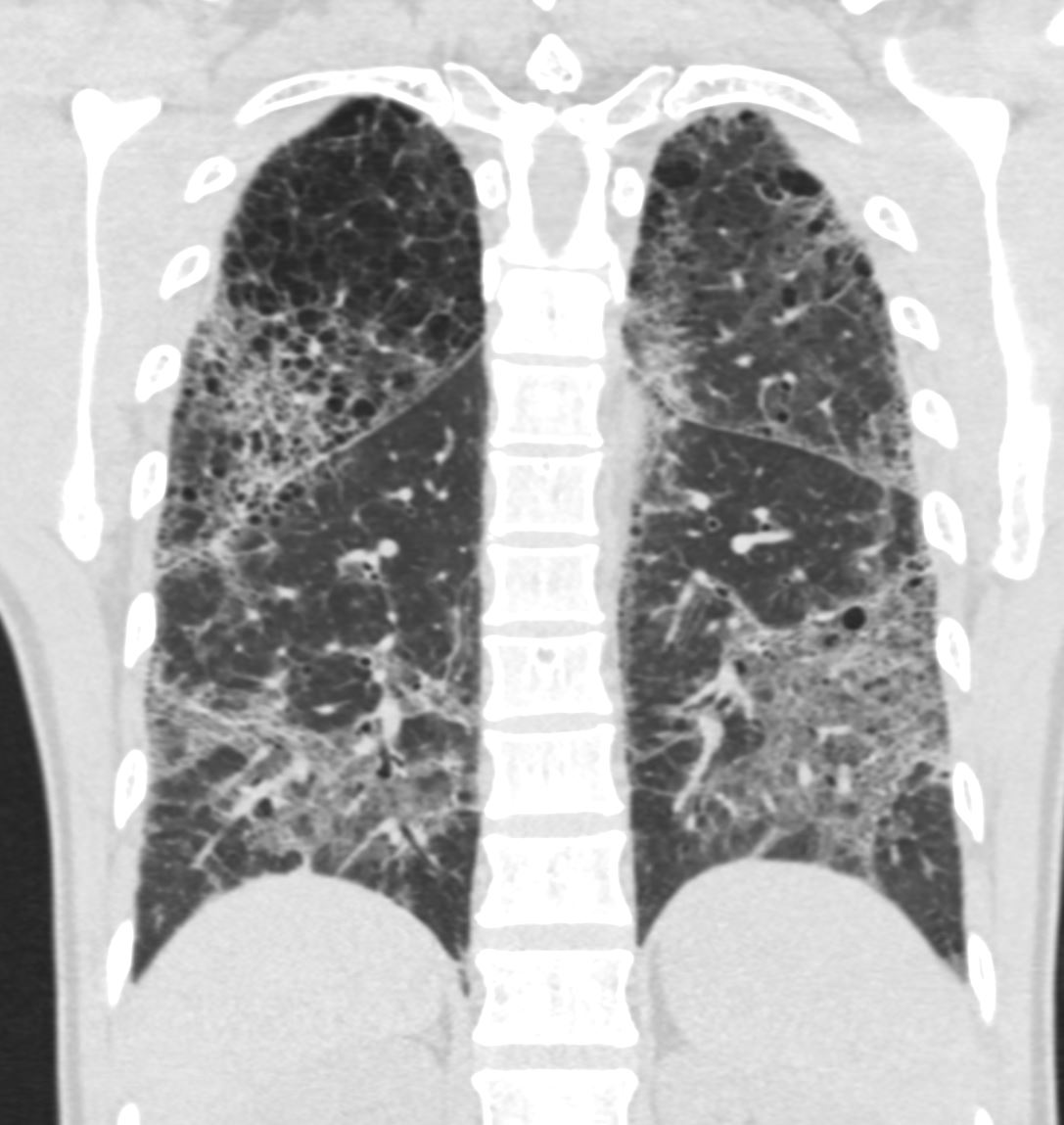
CT scan in shows multifocal regions of ground glass opacity in both the lower and upper lobes, mild progressive centrilobular emphysema but the presence of some small basilar cysts
LIP (lymphocytic interstitial pneumonia) is included in the radiological diffrential diagnosis
Ashley Davidoff MD
The CommonVein.net 139260 28Lu
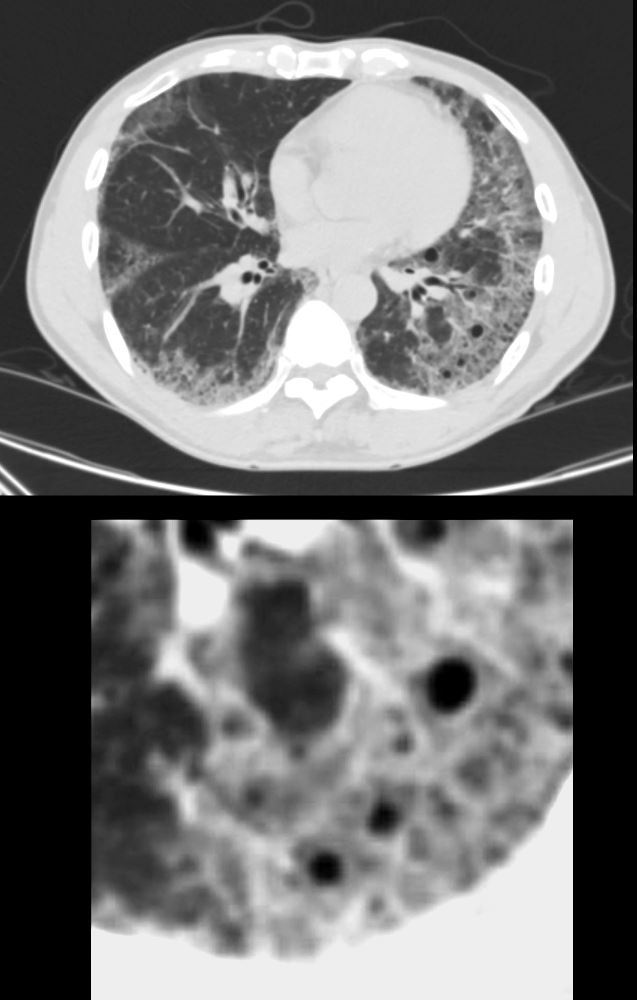
CT scan in the axial plane shows new multifocal regions of ground glass opacity thickening of the interlobular septa and the presence of new small basilar cysts
LIP (lymphocytic interstitial pneumonia) is included in the radiological differential diagnosis
Ashley Davidoff MD
The CommonVein.net 139252-01 28Lu
CT scan in coronal projection (a) shows multifocal regions of ground glass opacity in both the lower and upper lobes, associated with mild centrilobular emphysema. The right upper lobe changes are magnified in b and show a reticular pattern as a result of interlobular septal thickening (pink arrowheads in b) The cystic changes (teal blue arrowheads) are part of an underlying emphysematous process.
Ashley Davidoff MD
The CommonVein.net 139260c01L 28Lu
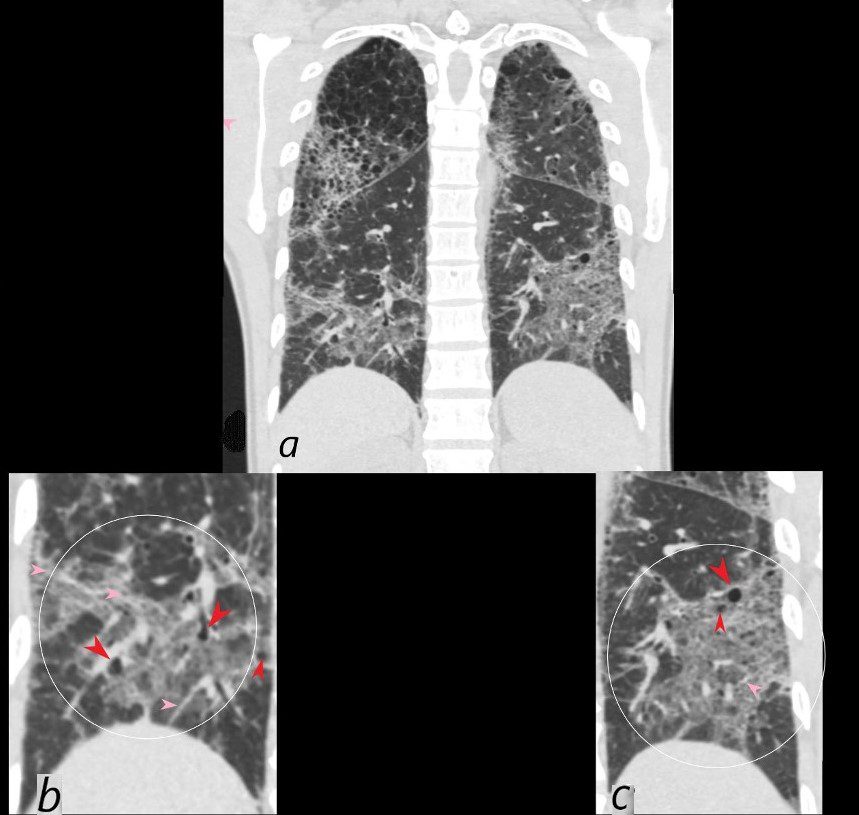
CT scan in coronal projection shows multifocal regions of ground glass opacity in both the lower and upper lobes, and mild centrilobular emphysema. The lower lobe changes are magnified in b and c and show multifocal regions of ground glass opacity , with thickening of the interlobular septa and the presence of new small bibasilar cysts (red arrowheads b and c)
LIP (lymphocytic interstitial pneumonia) is included in the radiological differential diagnosis
Ashley Davidoff MD
The CommonVein.net 139260c02L 28Lu
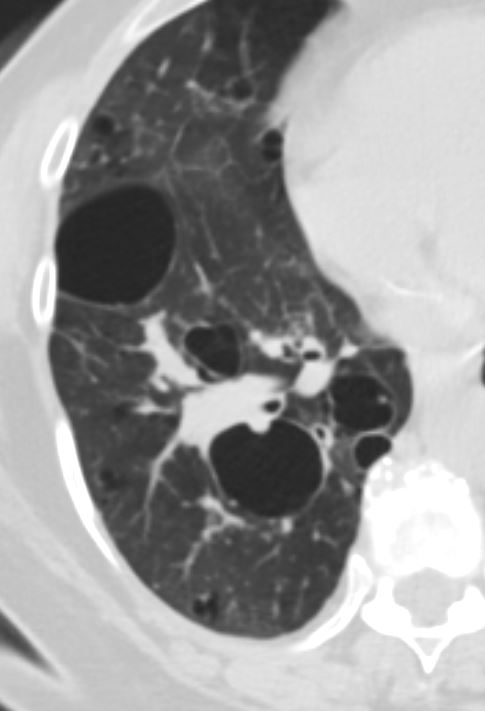
47 F SLE Sjogrens LIP vs Birt-Hogg-Dube basilar thin walled cysts lymphadenopathy
Subsegmental right lower lobe infiltrate
Ashley Davidoff TheCommonVein.net
47 F SLE Sjogrens LIP vs Birt-Hogg-Dube basilar thin walled cysts lymphadenopathy
Subsegmental right lower lobe infiltrate
Ashley Davidoff TheCommonVein.net
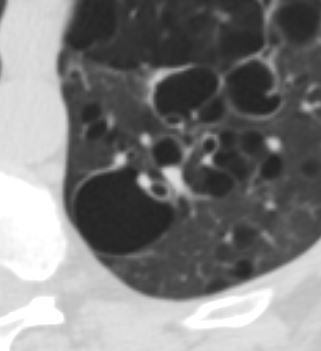
47 F SLE Sjogrens LIP vs Birt-Hogg-Dube basilar thin walled cysts lymphadenopathy
Subsegmental right lower lobe infiltrate
Ashley Davidoff TheCommonVein.net
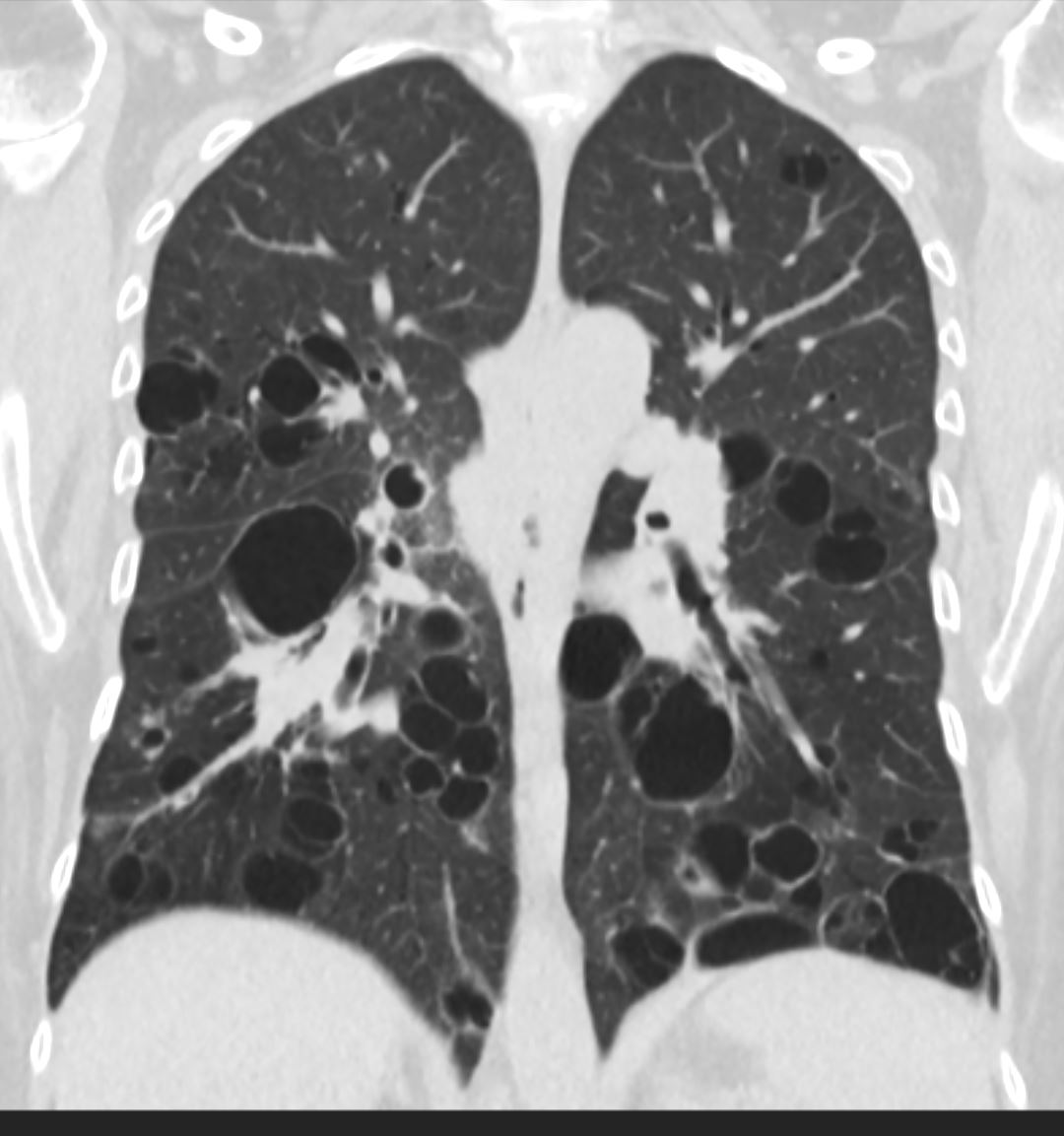
47 F SLE Sjogrens LIP vs Birt-Hogg-Dube basilar thin walled cysts lymphadenopathy
Subsegmental right lower lobe infiltrate
Ashley Davidoff TheCommonVein.net
| CT Finding | Description |
|---|---|
| Cysts and Nodules: | Development of cysts and nodules varying in size and distribution.
hypotheses perhaps bronchiolar obstruction similar to the pathogenesis of Langerhans cysts but many other hypotheses |
| Peribronchovascular and Perilymphatic Distribution: | Clustering of lymphocytic infiltrates around bronchovascular bundles and lymphatic vessels. |
| Interstitial Thickening: | Thickening of interlobular septa and bronchovascular bundles contributing to a reticular pattern. |
| Ground-Glass Centrilobular Nodules: | Centrilobular ground-glass nodules may be present. |
3D reconstruction of a CT scan shows the position of the lymphatic involvement of cellular infiltration along the bronchovascular bundle to the lower lobes
Ashley Davidoff MD TheCommonVein.net
32645a06b lymphatics
32645a06L01 lymphatic
| CT Finding | Description |
|---|---|
| Ground-Glass Opacities (GGOs): | Bilateral and diffuse opacities indicating partial airspace filling. |
| Interstitial Thickening: | Thickening of interlobular septa and bronchovascular bundles contributing to a reticular pattern. |
| Cysts and Nodules: | Development of cysts and nodules varying in size and distribution. |
| Ground-Glass Centrilobular Nodules: | Centrilobular ground-glass nodules may be present. |
| Pleural Effusion: | Pleural effusion is not common but mild effusion may be observed in some cases. |
Rare
- Typical onset at ages 40 – 70 years old but can occur at any age (Chest 2002;122:2150)
- Typically in patients with Sjogren’s syndrome
- lower lobe predominance
- GGO’s
- Can have MALT lymphoma
- and Amyloidosis Light chain deposition disease
- More common in women
- No association with smoking history
- Bilateral lower lobes of the lung but also mid lung zones
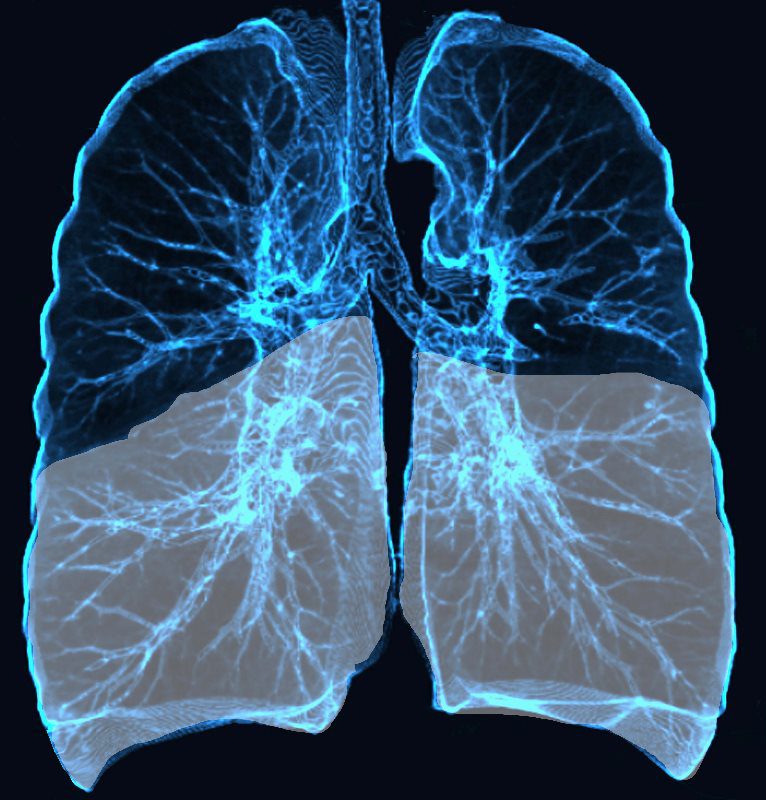
Lower Lobe distribution
Ashley Davidoff MD TheCommonvein.net lungs-0771
3D reconstruction of a CT scan shows the position of the lymphatic involvement of cellular infiltration along the bronchovascular bundle to the lower lobes
Ashley Davidoff MD TheCommonVein.net
32645a06b01lymphatics
Lower Lobes predominant Why?
- exact reason for this preference is
- not completely understood,
- One possible explanation
- lower lobes of the lungs are
- more dependent on gravity, which
- may affect the distribution of
- lymphocytes and
- other immune cells that are involved in the development of LIP.
- Gravity may cause these cells to accumulate more easily in the lower lobes, where
- blood flow and ventilation are also generally
- lower
- compared to the upper lobes.
- lymphatic drainage of the lower lobes
- less than the upper lobes, which could lead to the
- accumulation of lymphocytes in the lower lobes.
- certain infections or environmental exposures that may
- contribute to the development of LIP
- may also have a greater impact on the lower lobes of the lungs.
Disease Process
- infiltration of lymphocytes and plasma cells into the lung interstitium
- Pathogenic mechanisms of LIP are still unclear
- Has aspects of lymphoproliferative disease and lymphoid hyperplasia of polyclonal T or B cells (Chest 2002;122:2150)
- Although it may transform to lymphoma, especially MALT, the risk is lower than initially reported (Eur Respir J 2006;28:364)
- Associated with several systemic diseases and conditions
- Autoimmune (most common)
- Sjögren syndrome (SjS); 25% of LIP cases have SjS and 1% of SjS cases present with LIP
- Rheumatoid arthritis
- Systemic lupus erythematosus
- Polymyositis / dermatomyositis
- Hashimoto disease
- Hypothyroidism
- Castleman disease
- Myasthenia gravis
- Autoimmune hemolytic anemia
- Pernicious anemia
- Primary biliary cirrhosis
- Infection
- Human immunodeficiency virus (HIV)
- Epstein-Barr virus
- Human T cell lymphotropic virus type 1
- Legionella pneumonia
- Mycoplasma
- Chlamydia
- Tuberculosis
- Immunodeficiency
- Acquired immunodeficiency syndrome (AIDS); especially in children
- Monoclonal or polyclonal gammopathy
- Common variable immunodeficiency
- Idiopathic LIP accounts for 20% of cases (Eur Respir J 2006;28:364)
- Autoimmune (most common)
- Very slowly progressive respiratory symptoms
- Dyspnea on exertion
- Dry cough
- Systemic symptoms such as malaise, fever and weight loss
- Duration of the symptoms prior to diagnosis can exceed a year
- Bibasilar inspiratory crackles on chest auscultation
- Based on clinical, radiological and pathological findings (multidisciplinary diagnosis)
- No firm diagnostic criteria currently exist
- Dysproteinemia is often present
- Hypergammaglobulinemia is more common than hypogammaglobulinemia
- Restrictive pattern on pulmonary function tests
- Reduced forced vital capacity (FVC)
- Reduced diffusing capacity of the lung for carbon monoxide (DLCO)
- Chest radiography
- Bibasilar opacities with lower lobe predominance
- High resolution computed tomography (Eur J Radiol 2015;84:542, Respirology 2016;21:600)
- Ground glass opacity with / without consolidation with lower lobe predominance
- Cyst formation and thickening of bronchovascular bundle and interlobular septa are often present
- Cysts often remain even after resolution of symptoms
27 year old female with congenital HIV/AIDS and B cell Lymphoma


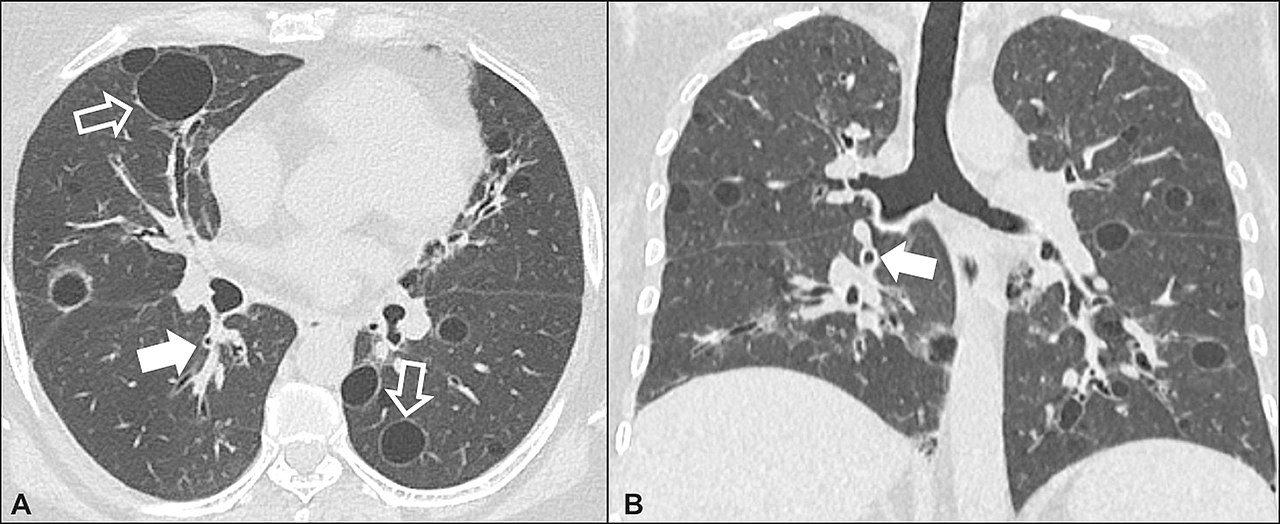
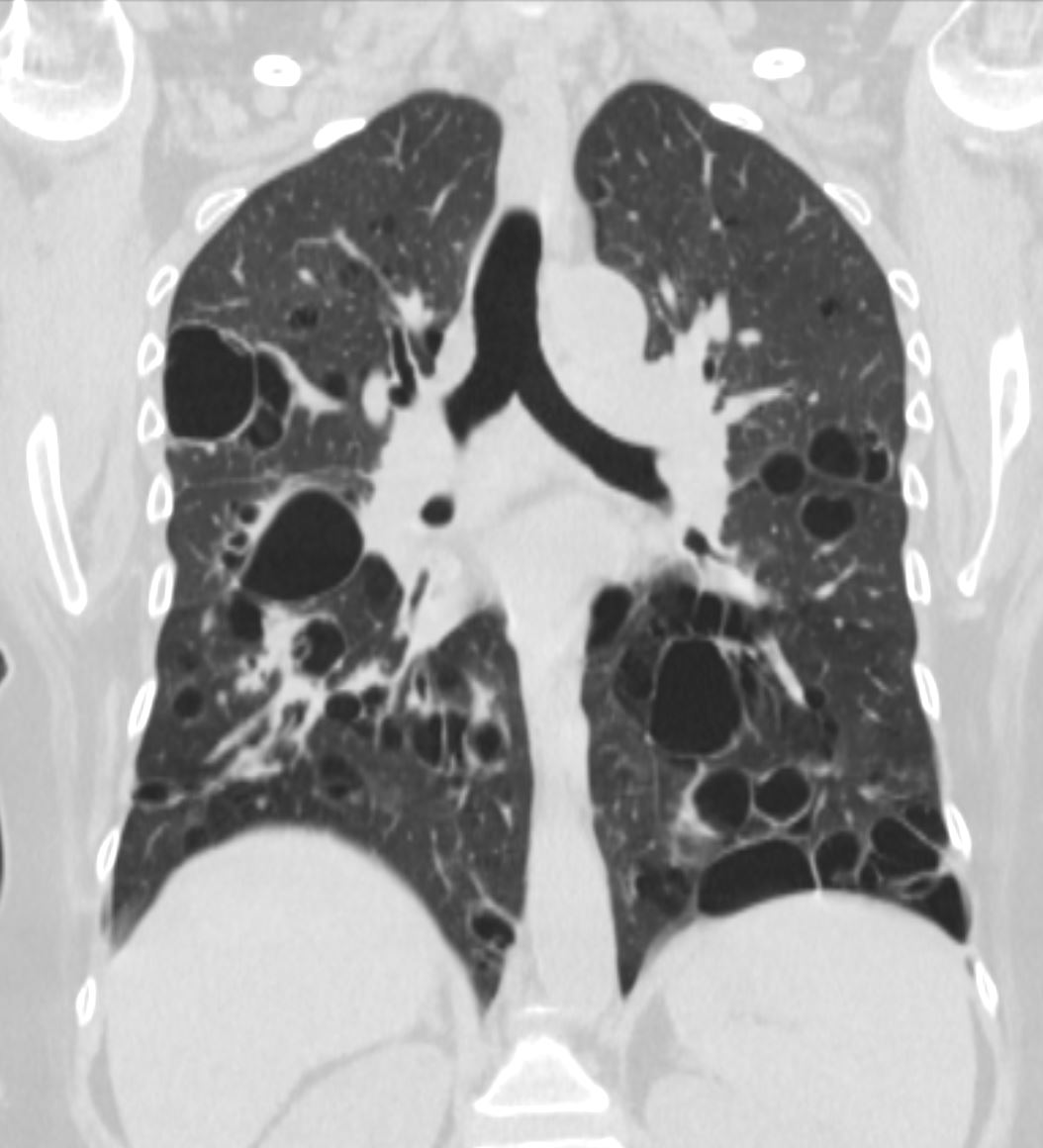
Lymphocytic interstitial pneumonia. A 62-year-old female patient with Sjögren’s syndrome. Axial high-resolution computed tomography scan of the chest (A) and coronal reformatting (B). In A, diffuse thickening of the bronchial walls (closed arrows), some ground-glass opacities and thin-walled cysts of varying sizes, with a diffuse, bilateral distribution (open arrows). In B, distribution predominantly in the lower fields.
Daniel Simões Oliveira et al
Radiologia Brasileira 51 (5): 321–327.
References and Links
Radiopaedia
Yoshikawa A, et al . Pathology Outlines.com
- TCV
-
- Usual interstitial pneumonia (UIP)
- Nonspecific interstitial pneumonia (NSIP)
- Cryptogenic organizing pneumonia (COP)
- Desquamative interstitial pneumonia (DIP)
- Respiratory bronchiolitis-interstitial lung disease (RB-ILD)
- Acute interstitial pneumonia (AIP)
- Lymphoid interstitial pneumonia (LIP)
- Idiopathic pleuroparenchymal fibroelastosis (PPFE)